
N Goalby chemrevise.org 1
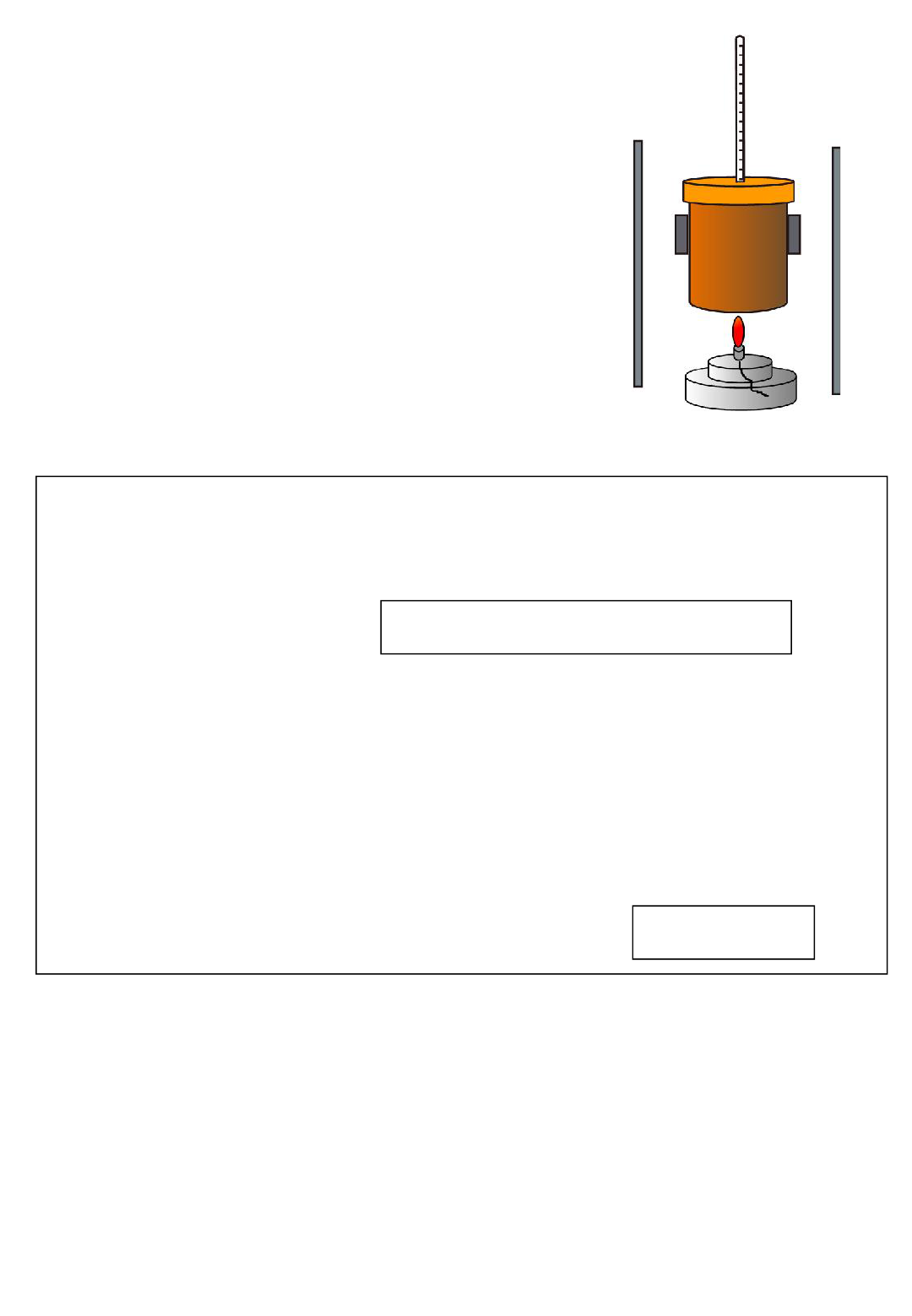
Potential errors in using a gas syringe
•gas escapes before bung inserted
•syringe sticks
• some gases like carbon dioxide or sulphur dioxide are
soluble in water so the true amount of gas is not
measured.
Using a gas syringe
If drawing a gas syringe make sure
you draw it with some
measurement markings on the
barrel to show measurements can
be made.
The volume of a gas depends on pressure
and temperature so when recording volume
it is important to note down the
temperature and pressure of the room.
Make sure you don’t leave gaps in
your diagram where gas could
escape
Gas syringes can be used for a variety of
experiments where the volume of a gas is
measured, possibly to work out moles of
gas or to follow reaction rates.
Moles of gas can be calculated from gas
volume (and temperature and pressure)
using ideal gas equation PV = nRT or using
the molar gas volume (1mol gas =24dm
3
at
room temperature and pressure
Measuring gas volumes
Alternatively gas volumes can be
measured ‘over water’ with an up-turned
measuring cylinder in a trough of water
Practical Guide EDEXCEL
This guide includes details about the core practicals for A-level chemistry. It also contains information about
other experiments that often occur in A-level examinations. You may be asked to describe these experiments in
details or be asked about reasons for doing individual steps.
You may be asked about other unfamiliar experiments but these will be using the skills and techniques that are
described in the following experiments.
Irritant - dilute acid and alkalis- wear googles
Corrosive- stronger acids and alkalis wear goggles
Flammable – keep away from naked flames
Toxic – wear gloves- avoid skin contact- wash hands after use
Oxidising- Keep away from flammable / easily oxidised materials
Hazardous substances in low
concentrations or amounts will
not pose the same risks as the
pure substance.
Safety and hazards

N Goalby chemrevise.org 2
Detailed method
1. Measure 30 cm
3
of 1 mol dm⁻
3
ethanoic acid and transfer
to a conical flask.
2. Attach conical flask to gas syringe or use collection over
water method (see previous page)
3. Measure the mass of a weighing bottle with approximately
0.05 g of calcium carbonate
4. Add the calcium carbonate to the conical flask- quickly
resealing the bung so no gas escapes
5. Measure the final total volume of gas
6. Reweigh the empty weighing bottle test tube from step 3
7. Repeat the experiment several more times, increasing the
mass of calcium carbonate by about 0.05 g each time.
Method for using a gas syringe to calculate the Mr of propanone
1. Extract 0.20 cm
3
of propanone into a hypodermic syringe and then measure the mass of this syringe
2. using hand protection, remove a gas syringe from the oven and note the volume of air already in the
barrel – about 5 cm
3.
3. inject the propanone through the self-seal cap into the barrel. The plunger will move straight away.
4. Put the gas syringe back into the oven.
5. Measure the mass of the empty hypodermic syringe immediately.
6. After a few minutes measure the volume of the gas in the gas syringe, record the temperature of the oven
shelf and the pressure of the room.
Example 1 : 0.150g of a volatile liquid was injected into a sealed gas syringe. The gas syringe was placed in an oven at
70
o
C at a pressure of 100kPa and a volume of 80cm
3
was measured. What is the Mr of the volatile liquid ? (R = 8.31)
moles = PV/RT
= 100 000 x 0.00008 / (8.31 x 343)
= 0.00281 mol
100 kPa = 100 000 Pa
80 cm
3
= 0.00008 m
3
Mr = mass/moles
= 0.15 / 0.00281
= 53.4 g mol
-1
Core Practical 1:Measure the molar volume of a gas
Mass of CaCO
3
in g
Volume of CO
2
in cm
3
Analysis
From the graph read the volume of CO
2
given off with 0.25 g CaCO
3
Work out the moles of CaCO
3
in 0.25g = 0.25/100.1 = 2.5 x 10
-3
Assume the moles of CO
2
= moles of CaCO
3
Work out molar volume of CO
2
= volume of CO
2
/ moles of CO
2

3
The water of crystallisation in calcium sulphate crystals can be
removed as water vapour by heating as shown in the following
equation.
CaSO
4
.xH
2
O(s) → CaSO
4
(s) + xH
2
O(g)
Method.
•Weigh an empty clean dry crucible and lid .
•Add 2g of hydrated calcium sulphate to the crucible and weigh
again
•Heat strongly with a Bunsen for a couple of minutes
•Allow to cool
•Weigh the crucible and contents again
•Heat crucible again and reweigh until you reach a constant mass (
do this to ensure reaction is complete).
Small amounts of the solid , such as
0.100 g, should not be used in this
experiment as the percentage
uncertainties in weighing will be too
high.
Large amounts of hydrated calcium sulphate, such as 50g,
should not be used in this experiment as the decomposition is
likely to be incomplete.
The lid improves the accuracy of the
experiment as it prevents loss of solid
from the crucible but should be loose
fitting to allow gas to escape.
The crucible needs to be dry otherwise a wet crucible would
give an inaccurate result. It would cause mass loss to be too
large as the water would be lost when heating.
Heating in a crucible
This method could be used for measuring mass loss in various
thermal decomposition reactions and also for mass gain when
reacting magnesium in oxygen.
N Goalby chemrevise.org
Example 2. 3.51 g of hydrated zinc sulphate were heated and 1.97 g of anhydrous zinc sulphate were
obtained. Use these data to calculate the value of the integer x in ZnSO
4
.xH
2
O
Calculate the mass of H
2
O = 3.51 – 1.97 = 1.54g
Calculate moles of
ZnSO
4
Calculate moles of
H
2
O
1.97
161.5
=
1.54
18
=
= 0.0122
=0.085
Calculate ratio of mole of
ZnSO
4
to H
2
O
0.0122
0.0122
= 0.085
0.0122
=1
=7
=
X = 7

N Goalby chemrevise.org 4
• Weigh the sample bottle containing the required mass of solid
on a 2 dp balance
• Transfer to beaker
• Reweigh empty sample bottle
• Record the difference in mass
• Add 100cm
3
of distilled water to the beaker. Use a glass rod to
stir to help dissolve the solid.
•Sometimes the substance may not dissolve well in cold water so
the beaker and its contents could be heated gently until all the
solid had dissolved.
• Pour solution into a 250cm
3
graduated flask via a funnel.
• Rinse beaker and funnel and add washings from the beaker
and glass rod to the volumetric flask.
• make up to the mark with distilled water using a dropping
pipette for last few drops.
• Invert flask several times to ensure uniform solution.
Making a solution
Alternatively the known mass of solid
in the weighing bottle could be
transferred to beaker, washed and
washings added to the beaker.
Remember to fill so the bottom of the
meniscus sits on the line on the neck of the
flask. With dark liquids like potassium
manganate it can be difficult to see the
meniscus.
Shake the volumetric flask thoroughly to
ensure a uniform concentration
Graduated/volumetric flask
A graduated flask has one mark on the neck which the level to
fill to get the accurate volume. Do not heat or put hot
solutions in the volumetric flask because the heat would cause
the flask to expand and the volume would then be incorrect.
Use a teat pipette to make up to the mark in
volumetric flask to ensure volume of solution
accurately measured and one doesn’t go over
the line
Diluting a solution
•Pipette 25cm
3
of original solution into a 250cm
3
volumetric
flask
•make up to the mark with distilled water using a dropping
pipette for last few drops.
• Invert flask several times to ensure uniform solution.
Using a volumetric pipette is more accurate
than a measuring cylinder because it has a
smaller uncertainty
Dilutions
Core practical 2. Make up a volumetric solution and carry out a simple acid–base titration
Measuring mass accurately:
In many experiments the best method for measuring mass is
1. Measure mass on 2 or 3d.p. balance of a weighing bottle
with the required quantity of solid in it
2. Empty mass into reaction vessel/flask
3. Reweigh the now empty weighing bottle
4. Subtract the mass of the empty weighing bottle from the
first reading to give exact of mass actually added.

N Goalby chemrevise.org 5
Titrations are done often to find out the concentration of one
substance by reacting it with another substance of known
concentration.
They are often done with neutralisation reactions, but can be
done with redox reactions.
However, the standard phrase: titrate solution A
with solution B means that A should be in the
conical flask and B should be in the burette.
One substance (generally the one we don’t know the
concentration) is put in the conical flask. It is measured
using a volumetric pipette.
The other substance is placed in the burette
burette
pipette
conical
flask
Using the pipette
Detailed Method for Titration
•rinse pipette with substance to go in it (often alkali).
•pipette 25 cm
3
of solution A into conical flask. The
volumetric pipette will have a mark on its neck to show
the level to fill to. The bottom of the meniscus should
sit on this line.
•touch surface of solution with pipette ( to ensure
correct amount is added). A small amount of solution
will be left in the pipette at this stage. The calibration
of the pipette will take into account this effect. It
should not be forced out.
Make sure bottom of
meniscus is on line on
neck of pipette
Titrations
A conical flask is used in preference to a beaker because
it is easier to swirl the mixture in a conical flask without
spilling the contents.
General Method
•rinse equipment (burette with acid, pipette with alkali, conical flask with distilled water)
•pipette 25 cm
3
of alkali into conical flask
•touch surface of alkali with pipette ( to ensure correct amount is added)
•adds acid solution from burette
•make sure the jet space in the burette is filled with acid
•add a few drops of indicator and refer to colour change at end point
•phenolphthalein [pink (alkali) to colourless (acid): end point pink colour just disappears] [use if NaOH is used]
•methyl orange [yellow (alkali) to red (acid): end point orange] [use if HCl is used]
•use a white tile underneath the flask to help observe the colour change
•add acid to alkali whilst swirling the mixture and add acid drop wise at end point
•note burette reading before and after addition of acid
•repeats titration until at least 2 concordant results are obtained- two readings within 0.1 of each other
Core Practical 3: Make up a volumetric solution and carry out a simple acid–base titration

N Goalby chemrevise.org
6
Using the burette
The burette should be rinsed out with substance that will be
put in it. If it is not rinsed out the acid or alkali added may be
diluted by residual water in the burette or may react with
substances left from a previous titration. This would lead to
the concentration of the substance being lowered and a larger
titre being delivered.
Don’t leave the funnel in the burette because small drops of
liquid may fall from the funnel during the titration leading to a
false burette reading (would give a lower titre volume)
make sure the jet space in the burette is filled with the
solution and air bubbles are removed.
If the jet space in the burette is not filled properly prior to commencing the
titration it will lead to errors if it then fills during the titration, leading to a
larger than expected titre reading.
Read the bottom of the meniscus on the burette
This is reading 9.00cm
3
Even though a burette has marking reading to 0.1cm
3
,
the burette readings
should always be given to 2dp either ending in 0.00 or 0.05. 0.05cm
3
is the
volume of 1 drop of solution delivered from a burette and so this is the
smallest difference in readings that can be measured. If the bottom of the
meniscus sits on a line it should end with a 0.00 as in the above example
9.00cm
3.
If the meniscus sits between two lines it should end 0.05. e.g. if the
bottom of the meniscus sits between the lines marked 9.1 and 9.2, you should
record 9.15
Add a few drops of indicator and refer to colour change
at end point
Adding indicator
phenolphthalein
If acid is added from the burette the colour change would
be pink (alkali) to colourless (acid): end point pink colour
just disappears [use with titrations using strong alkalis e.g.
NaOH ]
phenolphthalein
Alkali colour
phenolphthalein acid
colour
Methyl orange
Use a white tile underneath the flask to help
observe the colour change
Methyl orange is a suitable indicator for neutralisation
reactions where strong acids are used.
It is red in acid and yellow in alkali. It is orange at the end
point.
Methyl orange
Alkali colour
Methyl orange
acid colour
Methyl orange
end point
Indicators are generally weak acids so only add a few
drops of them. If too much is added they will affect
the titration result

N Goalby chemrevise.org 7
Add solution from burette whilst swirling the mixture and add drop-wise at end
point
note burette reading before and after addition of solution
repeats titration until at least 2 concordant results are
obtained- two readings within 0.1 of each other
Distilled water can be added to the conical flask during a titration to wash the
sides of the flask so that all the acid on the side is washed into the reaction
mixture to react with the alkali.
It does not affect the titration reading as water does not react with the reagents
or change the number of moles of acid added.
Only distilled water should be used to
wash out conical flasks between titrations
because it does not add any extra moles
of reagents
Recording results
•Results should be clearly recorded in a table
•Result should be recorded in full (i.e. both initial and final readings)
•Record titre volumes to 2dp (0.05 cm
3
)
Working out average titre results
Only make an average of the
concordant titre results
lf 2 or 3 values are within 0.10cm
3
and
therefore concordant or close then we
can say results are accurate and
repeatable and the titration technique
is good and consistent
Titration number 1 2 3
Initial burette reading (cm
3
) 0.50 2.50 1.55
Final burette reading (cm
3
) 24.50 27.00 25.95
Titre (cm
3
) 24.00 24.50 24.40
Average titre = (24.50+ 24.40)/2 =
24.45
Safety precautions
Acids and alkalis are corrosive
(at low concentrations acids are irritants)
Wear eye protection and gloves
If spilled immediately wash affected parts after spillage
If substance is unknown treat it as potentially toxic and wear
gloves.
A single titration could be flawed. Repeating allows for
anomalous titres to be spotted and discounted
Testing batches
In quality control it will be necessary to do titrations/testing
on several samples as the amount/concentration of the
chemical being tested may vary between samples.
Titrating mixtures
If titrating a mixture to work out the concentration of
an active ingredient it is necessary to consider if the
mixture contains other substances that have acid
base properties.
If they don’t have acid base properties we can titrate
with confidence.
Common Titration Equations
CH
3
CO
2
H + NaOH CH
3
CO
2
-
Na
+
+ H
2
O
H
2
SO
4
+ 2NaOH Na
2
SO
4
+2H
2
O
HCl + NaOH NaCl +H
2
O
NaHCO
3
+ HCl NaCl + CO
2
+ H
2
O
Na
2
CO
3
+ 2HCl 2NaCl + CO
2
+ H
2
O

N Goalby chemrevise.org 8
Calculating Apparatus Uncertainties
Each type of apparatus has a sensitivity uncertainty
•balance 0.001 g (if using a 3 d.p. balance)
•volumetric flask 0.1 cm
3
•25 cm
3
pipette 0.1 cm
3
•burette (start & end readings and end point ) 0.15 cm
3
Calculate the percentage error for each piece of equipment used by
% uncertainty = uncertainty x 100
Measurement made on apparatus
e.g. for burette
% uncertainty = 0.15/average titre result x100
To calculate the maximum total percentage apparatus uncertainty in the
final result add all the individual equipment uncertainties together.
Reducing uncertainties in a titration
Replacing measuring cylinders with pipettes or burettes which have
lower apparatus uncertainty will lower the error.
To reduce the uncertainty in a burette reading it is necessary to make
the titre a larger volume. This could be done by: increasing the volume
and concentration of the substance in the conical flask or by decreasing
the concentration of the substance in the burette.
To decrease the apparatus uncertainties
you can either decrease the sensitivity
uncertainty by using apparatus with a
greater resolution (finer scale divisions ) or
you can increase the size of the
measurement made.
If looking at a series of measurements in
an investigation, the experiments with
the smallest readings will have the
highest experimental uncertainties.
Reducing uncertainties in measuring mass
Using a more accurate balance or a larger mass will reduce the
uncertainty in weighing a solid
Weighing sample before and after addition and then
calculating difference will ensure a more accurate
measurement of the mass added.
Calculating the percentage difference between the actual
value and the calculated value
If we calculated an Mr of 203 and the real value is 214, then
the calculation is as follows:
Calculate difference 214-203 = 11
% = 11/214 x100
=5.41%
If the %uncertainty due to the apparatus < percentage
difference between the actual value and the calculated
value then there is a discrepancy in the result due to
other errors.
If the %uncertainty due to the apparatus > percentage
difference between the actual value and the calculated
value then there is no discrepancy and all errors in the
results can be explained by the sensitivity of the
equipment.
Uncertainty
In general, if uncertainty is not indicated
on apparatus, the following assumptions
are made:
For an analogue scale-
The uncertainty of a reading (one
judgement) is at least ±0.5 of the smallest
scale reading.
The uncertainty of a measurement (two
judgements) is at least ±1 of the smallest
scale reading.
- If the apparatus has a digital scale, the
uncertainty is the resolution of the
apparatus in each measurement
Readings and Measurements
Readings
the values found from a single
judgement when using a piece
of equipment
Measurements
the values taken as the
difference between the
judgements of two values
(e.g. using a burette in a
titration)
Uncertainty of a measurement using
a burette. If the burette used in the
titration had an uncertainty for each
reading of +/– 0.05 cm
3
then during a
titration two readings would be taken
so the uncertainty on the titre volume
would be +/– 0.10 cm
3
.

N Goalby chemrevise.org 9
Testing for halogenoalkanes method
• Arrange three test tubes in a row and add three drops of halogenoalkane in the sequence 1-chlorobutane, 1-
bromobutane, 1-iodobutane.
• Add 4 cm
3
of 0.02 M silver nitrate to each halogenoalkane.
• Without delay, put all three test tubes simultaneously in a hot water bath.
• Note the order in which precipitates appear
Comparing the rate of hydrolysis of halogenoalkanes reaction
Water is a poor nucleophile but it can react
slowly with haloalkanes in a substitution
reaction
Hydrolysis is defined as the splitting of a molecule ( in this case a
haloalkane) by a reaction with water
CH
3
CH
2
X + H
2
O CH
3
CH
2
OH + X
-
+ H
+
Aqueous silver nitrate is added to a haloalkane and the halide
leaving group combines with a silver ion to form a SILVER HALIDE
PRECIPITATE.
The precipitate only forms when the halide ion has left the
haloalkane and so the rate of formation of the precipitate can be
used to compare the reactivity of the different haloalkanes.
CH
3
CH
2
I + H
2
O CH
3
CH
2
OH + I
-
+ H
+
Ag
+
(aq)
+ I
-
(aq)
AgI
(s)
- yellow precipitate
The iodoalkane forms a precipitate with the
silver nitrate first as the C-I bond is weakest
and so it hydrolyses the quickest
The quicker the precipitate is formed, the faster the substitution
reaction and the more reactive the haloalkane
AgI
(s)
- yellow precipitate
AgBr
(s)
– cream precipitate
AgCl
(s)
– white precipitate
forms faster
The rate of these substitution reactions depends on the strength of the
C-X bond . The weaker the bond, the easier it is to break and the faster
the reaction.
Core practical 4: Investigation of the rates of hydrolysis of some halogenoalkanes

N Goalby chemrevise.org 10
Detailed Method: The partial oxidation of propan-1-ol
This experiment uses a limited quantity of oxidising agent (0.01 mol) and the product is distilled from the reaction
mixture immediately it is formed. In this way we hope to achieve a partial oxidation of propan-1-ol.
Place about 10 cm
3
of dilute sulphuric acid in a flask and add about 3g of potassium dichromate(VI) and 2 or 3
anti-bumping granules. Shake the contents of the flask until solution is complete (do not warm).
Add 1.5 cm
3
of propan-1-ol in drops from a dropping pipette, shaking the flask so as to mix the contents, and
then assemble distillation apparatus as shown below
Gently heat and slowly distil 2 cm
3
of liquid into a test tube, taking care that none of the reaction mixture
splashes over.
SAFETY
You must wear gloves when handling
solid potassium dichromate(Vl) since it is
highly toxic and a category 2 carcinogen;
it is also an irritant. Avoid inhaling any
dust.
Concentrated sulphuric acid is corrosive.
Partial Oxidation of Primary Alcohols
Reaction: primary alcohol aldehyde
Reagent: potassium dichromate (VI) solution and dilute sulphuric acid.
Conditions: (use a limited amount of dichromate) warm gently and distil out
the aldehyde as it forms:
C
O
C
H
H
H
H
Ethanal
Observation: the orange
dichromate ion (Cr
2
O
7
2-
) reduces
to the green Cr
3+
ion
propan-1-ol
propanal
+ [O]
+ H
2
O
CH
3
CH
2
CH
2
OH + [O] CH
3
CH
2
CHO + H
2
O
OH
+ [O]
O
+ H
2
O
C
O
C
H
H
C
HH
H
H
C O
H
H
H
C
H
H
C
H
H
H
Distillation
In general used as separation technique to separate an
organic product from its reacting mixture. Need to
collect the distillate of the approximate boiling point
range of the desired liquid.
Water in
Water
out
Liebig condenser
thermometer
Heat
Note the bulb of the thermometer should be
at the T junction connecting to the
condenser to measure the correct boiling
point
Note the water goes in the bottom of the
condenser to go against gravity. This allows more
efficient cooling and prevents back flow of water.
It’s important to be able to
draw and label this apparatus
accurately. Don’t draw lines
between flask, adaptor and
condenser.
Round
bottomed
flask
Electric heaters are often used to heat organic
chemicals. This is because organic chemicals are
normally highly flammable and could set on fire
with a naked flame.
Core practical 5. Oxidation of an alcohol

N Goalby chemrevise.org 11
Detailed method
Measure 5 cm
3
of water into a boiling tube. Add 6 g of sodium dichromate(VI), shake and set aside to dissolve.
Put about 1.5 cm
3
propan-1-ol into a 50 cm
3
round bottomed flask and add about 5 cm
3
of water and two or
three anti-bumping granules. Put a condenser on the flask for reflux, as shown in figure below.
Add 2 cm
3
of concentrated sulphuric acid down the condenser in drops from a dropping pipette. While the
mixture is still warm, start to add your sodium dichromate(VI) solution down the condenser in drops from a
dropping pipette. The energy released from the reaction should make the mixture boil. Add the solution a drop
at a time so that the mixture continues to boil without any external heating.
When all the sodium dichromate(VI) solution has been added, use a low Bunsen burner flame to keep the
mixture boiling for 10 minutes, not allowing any vapour to escape.
At the end of that time remove the Bunsen burner and arrange the apparatus for distillation. Gently distil 2-3
cm
3
of liquid into a test tube.
Reflux: Full Oxidation of Primary Alcohols
Reaction: primary alcohol carboxylic acid
Reagent: potassium/sodium dichromate(VI) solution and
sulphuric acid
Conditions: use an excess of dichromate, and heat under
reflux: (distil off product after the reaction has
finished)
C C
O
O H
H
C
H
H
H
H
Propanoic acid
propan-1-ol
Propanoic acid
Observation: the
orange dichromate
ion (Cr
2
O
7
2-
)
reduces to the
green Cr
3+
ion
CH
3
CH
2
CH
2
OH + 2[O] CH
3
CH
2
COOH + H
2
O
OH
+ 2[O]
+ H
2
O
O
OH
Reflux
Reflux is used when heating organic reaction mixtures for long periods. The
condenser prevents organic vapours from escaping by condensing them back
to liquids. The reactant vapours of volatile compound are condensed and
returned to the reaction mixture.
Never seal the end of the condenser as the build up of gas
pressure could cause the apparatus to explode. This is true of any
apparatus where volatile liquids are heated including the
distillation set up
Water in
Water out
Heat
Anti-bumping granules are added to the flask in both distillation
and reflux to prevent vigorous, uneven boiling by making small
bubbles form instead of large bubbles
It’s important to be able to draw and label this apparatus
accurately.
• Don’t draw lines between flask and condenser.
• Don’t have top of condenser sealed
• Condenser must have outer tube for water that is sealed
at top and bottom
• Condenser must have two openings for water in and out
that are open
Round
bottomed
flask
condenser

N Goalby chemrevise.org 12
Fractional Distillation: In the laboratory
• Heat the flask, with a Bunsen burner or electric mantle
• This causes vapours of all the components in the mixture
to be produced.
• Vapours pass up the fractionating column.
• The vapour of the substance with the lower boiling point
reaches the top of the fractionating column first.
• The thermometer should be at or below the boiling point
of the most volatile substance.
• The vapours with higher boiling points condense back
into the flask.
• Only the most volatile vapour passes into the condenser.
• The condenser cools the vapours and condenses to a
liquid and is collected.
Fractional distillation is used to
separate liquids with different
boiling points
flask
condenser
fractionating column

N Goalby chemrevise.org 13
The drying agent should
•be insoluble in the organic liquid
• not react with the organic liquid
• Put the distillate of impure product into a separating funnel
• wash product by adding either
• sodium hydrogencarbonate solution , shaking and
releasing the pressure from CO
2
produced.
• Saturated sodium chloride solution
•Allow the layers to separate in the funnel, and then run and
discard the aqueous layer.
•Run the organic layer into a clean, dry conical flask and add three
spatula loads of drying agent (e.g. anhydrous sodium sulphate,
calcium chloride) to dry the organic liquid. When dry the organic
liquid should appear clear.
• Carefully decant the liquid into the distillation flask
•Distill to collect pure product
Sodium hydrogencarbonate will neutralise
any remaining reactant acid.
Sodium chloride will help separate the
organic layer from the aqueous layer
Purifying an organic liquid
Separating funnel
General method
The layer with lower density will be the
upper layer. This is usually the organic layer
Decant means carefully pour off organic
liquid leaving the drying agent in the
conical flask
Distillation
In general used as separation technique to separate an
organic product from its reacting mixture. Need to
collect the distillate of the approximate boiling point
range of the desired liquid.
Water in
Water
out
Liebig condenser
thermometer
Heat
Note the bulb of the thermometer should be
at the T junction connecting to the
condenser to measure the correct boiling
point
Note the water goes in the bottom of the
condenser to go against gravity. This allows more
efficient cooling and prevents back flow of water.
It’s important to be able to
draw and label this apparatus
accurately. Don’t draw lines
between flask, adaptor and
condenser.
Round
bottomed
flask
Electric heaters are often used to heat organic
chemicals. This is because organic chemicals are
normally highly flammable and could set on fire
with a naked flame.

N Goalby chemrevise.org 14
1. Measure 8 cm
3
of 2-methylpropan-2-ol in a measuring cylinder and measure its mass.
2. Pour the 2-methylpropan-2-ol into a separating funnel, and reweigh the measuring cylinder to find the
mass of the 2-methylpropan-2-ol used.
3. In a fume cupboard, add 20 cm
3
of concentrated hydrochloric acid to the separating funnel, in portions
of 3cm
3
. After each portion, stopper the flask and invert it several times . Open the tap when doing this
to release the pressure.
4. Allow the separating funnel to stand in the fume cupboard for about 20 minutes. Gently shake it at
intervals.
5. After 20 minutes, allow the layers to separate in the funnel. Open the tap and remove the lower
aqueous layer. Dispose of this layer.
6. Add sodium hydrogencarbonate solution in 2 cm
3
portions to the separating funnel. This neutralises any
remaining acid. Shake the funnel after each addition, and release the pressure. Continue until no more
bubbles of CO
2
are seen.
7. Allow the layers to separate in the funnel. Again remove and pour away the lower aqueous layer. Run
off the organic layer into a clean conical flask and add two spatulas of anhydrous sodium sulfate.
Stopper the flask, shake the contents and allow this to stand until the liquid becomes clear. This step
dries the organic liquid.
8. Decant the liquid into a weighed clean distillation flask.
9. Distil the liquid by holding a 250ml beaker half-full of boiled water around the flask using standard
distillation set up. Collect the liquid that distils in the range 47-53
o
C.
10. Measure the mass of the 2-chloro-2-methylpropane collected.
Detailed method for preparing and purifying a halogenoalkane from an alcohol
a) Pour about 20 cm
3
of cyclohexanol into a weighed 50 cm
3
pear-shaped flask. Reweigh the flask and record
the mass of cyclohexanol.
b) Using a plastic graduated dropping pipette, carefully and with frequent shaking, add to the flask
approximately 8 cm
3
of concentrated phosphoric acid.
c ) Add a few anti-bumping granules to the flask and assemble the distillation apparatus, so that the contents
of the flask may be distilled. Heat the flask gently, distilling over any liquid which boils below 100 °C.
d) Pour the distillate into a separating funnel and add 50 cm
3
of saturated sodium chloride solution. Shake
the mixture and allow the two layers to separate.
e) run off the lower layer into a beaker and then transfer the upper layer, which contains the crude
cyclohexene, into a small conical flask.
f) Add a few lumps of anhydrous calcium chloride or anhydrous sodium sulfate(VI) or anhydrous magnesium
sulfate to the crude cyclohexene to remove water. Stopper the flask, shake the contents and allow this to
stand until the liquid becomes clear.
g) Decant the liquid into a clean, dry, weighed sample container.
h) Reweigh the container, calculate the mass of dry cyclohexene produced
Detailed method for preparing and purifying Cyclohexene from cyclohexanol
OH
+H
2
O
Conc
H
3
PO
4
CORE PRACTICAL 6: Chlorination of 2-methylpropan-2-ol using concentrated hydrochloric acid

N Goalby chemrevise.org 15
Detailed Method for Preparing and Purifying an Ester
Propyl ethanoate can be made in the laboratory from propan-
1-ol and ethanoic acid.
The equation for the reaction is
CH
3
COOH + CH
3
CH
2
CH
2
OH CH
3
COOCH
2
CH
2
CH
3
+ H
2
O
Procedure
1. Propan-1-ol (50 cm
3
) and ethanoic acid (50 cm
3
) are mixed
thoroughly in a 250 cm
3
round-bottomed flask.
2. Concentrated sulfuric acid (10 cm
3
) is added drop by drop
to the mixture, keeping the contents of the flask well-shaken
and cooled in an ice-water bath.
3. When the acid has all been added, a reflux condenser is
fitted to the flask and the mixture gently boiled over an
electric heating mantle for about 30 minutes.
4. The mixture is cooled, and the apparatus rearranged for
distillation. The crude ester (about 60 cm
3
) is distilled off.
5. The distillate is placed in a separating funnel and shaken
with about half its volume of 30% sodium carbonate solution,
with the pressure being released at intervals. The lower
aqueous layer is then discarded.
6. The crude ester is shaken in a separating funnel with about
half its volume of 50% calcium chloride solution, which
removes unreacted alcohol. The lower layer is discarded.
7. The ester is run into a clean, dry flask containing some
anhydrous calcium chloride and swirled.
8. The ester is filtered into a clean, dry flask, with a few anti-
bumping granules, and distilled. The fraction boiling between
100°C and 103°C is collected.
Sulfuric acid is a catalyst
Adding conc H
2
SO
4
is an exothermic reaction- to
prevent uncontrolled boiling over add drop by drop
and cool
In reflux the reactant vapours of volatile compound
are condensed and returned to the reaction mixture.
The reaction is slow so it is heated for 30 minutes
The electric heating mantle allows for controlled
heating and stops flammable vapour lighting
Sodium carbonate reacts with unreacted acid and
remaining catalyst still present after distillation.
The reaction produces CO
2
so the pressure of gas
needs to be released.
The upper layer is organic because it has a lower
density than water
Calcium chloride is a drying agent. The liquid will
appear clear when dry.
Anti-bumping granules are added to the prevent
vigorous, uneven boiling by making small bubbles
form instead of large bubbles
Measuring boiling point
Purity of liquid can be determined by measuring a boiling point. This can be done
in a distillation set up or by simply boiling a tube of the sample in an heating oil
bath. If the liquid is pure it will have the boiling point referred to in data books. If
impure the boiling point tends to be higher than the pure liquid
To get a correct measure of
boiling point the
thermometer should be
above the level of the surface
of the boiling liquid and be
measuring the temperature
of the saturated vapour.
Pressure should be noted as changing pressure can change the boiling point of
a liquid
Measuring boiling point is not the most accurate method of identifying a
substance as several substances may have the same boiling point.

N Goalby chemrevise.org 16
Testing for Ammonium ions (NH
4
+
)
a) Place about 10 drops of 0.1 mol dm
–3
ammonium chloride in a test tube.
b) Add about 10 drops of 0.4 mol dm
–3
sodium hydroxide solution. Shake the mixture.
c) Warm the mixture in the test tube gently using a water bath.
d) Test the fumes released from the mixture by holding a piece of damp red litmus
paper in the mouth of the test tube.
Method: adding dilute sodium hydroxide
a) Place about 10 drops of 0.1 mol dm
–3
metal ion solution in a test tube.
b) Add about 10 drops of 0.6 mol dm
–3
sodium hydroxide solution, mixing well.
c) Continue to add sodium hydroxide solution, dropwise with gentle shaking, until in excess
This test can be used
on group 2 metal
ions and transition
metal ions.
Results for Group 2
Magnesium hydroxide is classed as insoluble in water and
will appear as a white precipitate.
Simplest Ionic Equation for formation of Mg(OH)
2
(s)
Mg
2+
(aq) + 2OH
-
(aq) Mg(OH)
2
(s).
A suspension of magnesium hydroxide in water
will appear slightly alkaline (pH 9) so some
hydroxide ions must therefore have been
produced by a very slight dissolving.
Calcium hydroxide is classed as partially soluble in water and
will appear as a white precipitate (it may need more sodium
hydroxide to be added before it appears compared to a
magnesium solution.)
Simplest Ionic Equation for formation of Ca(OH)
2
(s)
Ca
2+
(aq) + 2OH
-
(aq) Ca(OH)
2
(s).
A suspension of calcium hydroxide in water will
appear more alkaline (pH 11) than magnesium
hydroxide as it is more soluble so there will be
more hydroxide ions present in solution.
The results in this test are an application of the trend that group II hydroxides become more soluble down the group.
Strontium and barium salts will not form a hydroxide precipitate on addition of
sodium hydroxide due to their high solubility. The solutions will be highly alkaline
Results for transition metals
Copper(II) solutions form a blue ppt,
Cobalt(II) solutions form a blue ppt,
iron (II) solutions form a green ppt
iron (III) solutions form a brown ppt
Chromium (III) solutions form a green
ppt which dissolves in excess to form a
green solution
[Cu(H
2
O)
6
]
2+
(aq)
+ 2OH
-
(aq)
Cu(H
2
O)
4
(OH)
2 (s)
+ 2H
2
O
(l)
[Fe(H
2
O)
6
]
2+
(aq)
+ 2OH
-
(aq)
Fe(H
2
O)
4
(OH)
2 (s)
+ 2H
2
O
(l)
[Fe(H
2
O)
6
]
3+
(aq)
+ 3OH
-
(aq)
Fe(H
2
O)
3
(OH)
3 (s)
+ 3H
2
O
(l)
Results: alkaline
ammonia gas is released
which turns the red
litmus paper blue
CORE PRACTICAL 7 + 15: Analysis of some inorganic unknowns
Testing for cations
Lithium : Scarlet red
Sodium : Yellow
Potassium : lilac
Rubidium : red
Caesium: blue
Magnesium: no flame colour (energy emitted of a
wavelength outside visible spectrum)
Calcium: brick red
Strontium: red
Barium: apple green
Method
Use a nichrome wire ( nichrome is an unreactive metal and
will not give out any flame colour)
Clean the wire by dipping in concentrated hydrochloric acid
and then heating in Bunsen flame
If the sample is not powdered then grind it up.
Dip wire in solid and put in Bunsen flame and observe flame
Flame tests
[Co(H
2
O)
6
]
2+
(aq)
+ 2OH
-
(aq)
Co(H
2
O)
4
(OH)
2 (s)
+ 2H
2
O
(l)
[Cr(H
2
O)
6
]
3+
(aq)
+ 3OH
-
(aq)
Cr(H
2
O)
3
(OH)
3 (s)
+ 3H
2
O
(l)
Cr(H
2
O)
3
(OH)
3 (s)
+ 3OH
-
(aq )
[Cr(OH)
6
]
3-
(aq)
+ 3H
2
O
(l)

N Goalby chemrevise.org 17
Testing for presence of a sulfate ion
BaCl
2
solution acidified with hydrochloric acid is used as a reagent to test for
sulphate ions.
If acidified Barium Chloride is added to a solution that contains sulfate ions a white
precipitate of Barium Sulfate forms.
Simplest ionic equation
Ba
2+
(aq) + SO
4
2-
(aq) BaSO
4
(s).
2HCl + Na
2
CO
3
2NaCl + H
2
O + CO
2
Fizzing due to CO
2
would be observed if a carbonate was present.
Other anions should give a negative
result which is no precipitate
forming.
The hydrochloric acid is needed to react with carbonate impurities that are often found in salts which
would form a white Barium carbonate precipitate and so give a false result. You could not used
sulphuric acid because it contains sulphate ions and so would give a false positive result.
Testing for presence of halide ions with silver nitrate.
This reaction is used as a test to identify which halide ion is
present. The test solution is made acidic with nitric acid, and
then Silver nitrate solution is added dropwise.
The role of nitric acid is to react with any
carbonates present to prevent formation of the
precipitate Ag
2
CO
3
. This would mask the desired
observations
2 HNO
3
+ Na
2
CO
3
2 NaNO
3
+ H
2
O + CO
2
Fluorides produce no precipitate
Chlorides produce a white precipitate
Ag
+
(aq) + Cl
-
(aq) AgCl(s)
Bromides produce a cream precipitate
Ag
+
(aq) + Br
-
(aq) AgBr(s)
Iodides produce a pale yellow precipitate
Ag
+
(aq) + I
-
(aq) AgI(s)
The silver halide precipitates can be treated with ammonia solution to help differentiate between them
if the colours look similar:
Silver chloride dissolves in dilute ammonia to form a complex ion
AgCl(s) + 2NH
3
(aq) [Ag(NH
3
)
2
]
+
(aq) + Cl
-
(aq)
Colourless solution
Silver bromide dissolves in concentrated ammonia to form a complex ion
AgBr(s) + 2NH
3
(aq) [Ag(NH
3
)
2
]
+
(aq) + Br
-
(aq)
Colourless solution
Silver iodide does not react with ammonia – it is too insoluble.
Fizzing due to CO
2
would be
observed if a carbonate was
present
Testing for presence of carbonate ions
Add any dilute acid and observe effervescence.
Bubble gas through limewater to test for CO
2
– will turn limewater cloudy
2HCl + Na
2
CO
3
2NaCl + H
2
O + CO
2
Testing for presence of a hydroxide ions
Alkaline hydroxide ions will turn red litmus paper blue
CORE PRACTICAL 7+15: Analysis of some inorganic unknowns
Testing for anions: – Group 7 (halide ions), OH
–
, CO
3
2–
, SO
4
2–

Reactions of halide salts with concentrated sulphuric acid.
Explanation of differing reducing power of halides
A reducing agent donates electrons.
The reducing power of the halides increases down group 7
They have a greater tendency to donate electrons.
This is because as the ions get bigger it is easier for the outer
electrons to be given away as the pull from the nucleus on them
becomes smaller.
The Halides show increasing power as reducing
agents as one goes down the group. This can be
clearly demonstrated in the various reactions of
the solid halides with concentrated sulphuric
acid.
Know the equations and observations of these
reactions very well.
F
-
and Cl
-
ions are not strong enough reducing agents to reduce the S in H
2
SO
4
. No redox
reactions occur. Only acid-base reactions occur.
Fluoride and Chloride
NaF(s) + H
2
SO
4
(l) NaHSO
4
(s) + HF(g)
Observations: White steamy fumes of HF are evolved.
NaCl(s) + H
2
SO
4
(l) NaHSO
4
(s) + HCl(g)
Observations: White steamy fumes of HCl are evolved.
These are acid –base reactions and not
redox reactions. H
2
SO
4
plays the role of
an acid (proton donor).
Br- ions are stronger reducing agents than Cl- and F- and after the initial acid-base
reaction reduce the Sulphur in H
2
SO
4
from +6 to + 4 in SO
2
Bromide
Acid- base step: NaBr(s) + H
2
SO
4
(l) NaHSO
4
(s) + HBr(g)
Redox step: 2HBr + H
2
SO
4
Br
2
(g) + SO
2
(g) + 2H
2
O(l)
Observations: White steamy fumes of
HBr are evolved.
Red fumes of Bromine are also evolved
and a colourless, acidic gas SO
2
Ox ½ equation 2Br
-
Br
2
+ 2e
-
Re ½ equation H
2
SO
4
+ 2 H
+
+ 2 e
-
SO
2
+ 2 H
2
O
Iodide
I- ions are the strongest halide reducing agents. They can reduce the Sulphur from +6 in
H
2
SO
4
to + 4 in SO
2
, to 0 in S and -2 in H
2
S.
NaI(s) + H
2
SO
4
(l) NaHSO
4
(s) + HI(g)
2HI + H
2
SO
4
I
2
(s) + SO
2
(g) + 2H
2
O(l)
6HI + H
2
SO
4
3 I
2
+ S (s) + 4 H
2
O (l)
8HI + H
2
SO
4
4I
2
(s) + H
2
S(g) + 4H
2
O(l)
Observations:
White steamy fumes of HI are evolved.
Black solid and purple fumes of Iodine are also
evolved
A colourless, acidic gas SO
2
A yellow solid of Sulphur
H
2
S (Hydrogen Sulphide), a gas with a bad egg
smell,
Ox ½ equation 2I
-
I
2
+ 2e
-
Re ½ equation H
2
SO
4
+ 2 H
+
+ 2 e
-
SO
2
+ 2 H
2
O
Re ½ equation H
2
SO
4
+ 6 H
+
+ 6 e
-
S + 4 H
2
O
Re ½ equation H
2
SO
4
+ 8 H
+
+ 8 e
-
H
2
S + 4 H
2
O
Often in exam questions these redox reactions are
worked out after first making the half-equations
Reduction product = sulphur dioxide
Note the H
2
SO
4
plays the role of acid in the first step producing HBr and then
acts as an oxidising agent in the second redox step.
Note the H
2
SO
4
plays the role of acid in the first step producing HI and then
acts as an oxidising agent in the three redox steps
Reduction products = sulphur dioxide, sulphur
and hydrogen sulphide
18
N Goalby chemrevise.org

N Goalby chemrevise.org 19
More on Insoluble salts and Precipitation reactions
Insoluble salts can be made by mixing appropriate solutions of ions so that a precipitate is formed
Barium nitrate (aq) + sodium sulfate (aq) Barium Sulfate (s) + sodium nitrate (aq)
These are called precipitation reactions. A precipitate is a solid
When making an insoluble salt, normally the salt would be removed by filtration, washed with distilled
water to remove soluble impurities and then dried on filter paper
Writing Ionic equations for precipitation reactions
We usually write ionic equations to show precipitation
reactions. Ionic equations only show the ions that are
reacting and leave out spectator ions.
Spectator ions are ions that are
• Not changing state
• Not changing oxidation number
Ba(NO
3
)
2
(aq) + Na
2
SO
4
(aq) BaSO
4
(s) + 2 NaNO
3
(aq)
Take full equation
Separate (aq) solutions into
ions
Ba
2+
(aq)
+ 2NO
3
-
(aq)
+ 2Na
+
(aq)
+ SO
4
2-
(aq)
BaSO
4(s)
+ 2 Na
+
(aq)
+ 2NO
3
-
(aq)
Cancel out spectator ions leaving
the simplest ionic equation
Ba
2+
(aq) + SO
4
2-
(aq) BaSO
4
(s).
There are some common rules for solubility of salts. No syllabus requires these to be learnt but a good
chemist does know them.
Soluble salts Insoluble salts
All sodium, potassium and ammonium salts
All nitrates
Most chlorides, bromides, iodides
Silver, lead chlorides, bromides iodides
Most
sulfates
Lead
sulfate strontium and barium sulfate
Sodium, potassium and ammonium
carbonates
Most other carbonates
Sodium, potassium and ammonium
hydroxides
Most other hydroxides
Filter
funnel
Filter
paper
residue
filtrate
This is gravitational filtration. Use
if small amounts of solid are
formed.
Buchner flask (has
thicker glass walls than
a normal flask to cope
with the vacuum )
Filter paper
This is vacuum filtration. The apparatus is
connected to a water pump which will
produce a vacuum. Use if larger amounts of
solid are formed.
Air outlet to
water pump
Buchner
funnel
For both types of filtration apparatus AQA expect filter paper to be drawn on the diagram
Filtration

N Goalby chemrevise.org 20
Tollen's reagent method
Place 1 cm
3
of silver nitrate solution in each of two clean boiling tubes.
Then add one drop of sodium hydroxide solution to form a precipitate of
silver oxide. Add ammonia solution dropwise until a clear, colourless
solution is formed. Add a few drops of the unknown and leave in the
water bath for a few minutes.
Functional group tests for an Aldehyde
Tollen’s Reagent
Reagent: Tollen’s Reagent formed by mixing aqueous ammonia and silver
nitrate. The active substance is the complex ion of [Ag(NH
3
)
2
]
+
.
Conditions: heat gently
Reaction: aldehydes only are oxidised by Tollen’s reagent into a carboxylic
acid and the silver(I) ions are reduced to silver atoms
Observation: with aldehydes, a silver mirror forms coating the inside of the
test tube. Ketones result in no change.
CH
3
CHO + 2Ag
+
+ H
2
O CH
3
COOH + 2Ag + 2H
+
Fehling's solution method
Place 1 cm
3
of Fehling's A into each of
two boiling tubes, and then add Fehling's
B until the blue precipitate redissolves.
Add a few drops of the unknown and
leave in the water bath for a few
minutes.
Reagent: Fehling’s Solution containing blue Cu
2+
ions.
Conditions: heat gently
Reaction: aldehydes only are oxidised by Fehling’s Solution
into a carboxylic acid and the copper (II) ions are
reduced to copper(I) oxide .
Observation: Aldehydes :Blue Cu
2+
ions in solution change
to a red precipitate of Cu
2
O. Ketones do not react
Fehling’s solution
CH
3
CHO + 2Cu
2+
+ 2H
2
O CH
3
COOH + Cu
2
O + 4H
+
Functional group test for an Alkene
To 0.5 cm
3
of bromine water in a test tube add a few drops of the unknown and shake.
Observation: alkenes should decolourise bromine water
Tests for alcohol, aldehyde, alkene and carboxylic acid
CORE PRACTICAL 7+15: Analysis of some organic unknowns
The melting point of the crystal formed can be used to help
identify which carbonyl was used. Take the melting point of
orange crystals product from 2,4-DNP. Compare melting point
with known values in database
Reaction with 2,4-dinitro phenylhydrazine
2,4-DNP reacts with both aldehydes and ketones. The product
is an orange precipitate, It can be used as a test for a carbonyl
group in a compound.
Use 2,4-DNP to identify if the compound is a
carbonyl. Then to differentiate an aldehyde
from a ketone use Tollen’s reagent.

N Goalby chemrevise.org 21
Summary of Identification of Functional Groups by test-tube reactions
Functional group test for a Carboxylic acid
To 0.5 cm
3
of your unknown solution in a test tube add a
small amount of sodium carbonate solid and observe.
Result carboxylic acids will fizz with sodium carbonate due
to CO
2
produced
The presence of a carboxylic acid can be tested by
addition of sodium carbonate. It will fizz and produce
carbon dioxide
2CH
3
CO
2
H + Na
2
CO
3
2CH
3
CO
2
-
Na
+
+ H
2
O + CO
2
Reaction of carbonyls with iodine in presence of alkali
Reagents: Iodine and sodium hydroxide
Conditions: warm very gently
Only carbonyls with a methyl group next to the
C=O bond will do this reaction. Ethanal is the
only aldehyde that reacts. More commonly is
methyl ketones.
CH
3
C
O
H
The product CHI
3
is a yellow crystalline
precipitate with an antiseptic smell
This reaction is called the Iodoform test
Functional group Reagent Result
Alkene Bromine water Orange colour
decolourises
Alcohols + carboxylic acids PCl
5
Misty fumes of HCl
produced
Alcohols, phenols,
carboxylic acids
Sodium metal Efferevesence due to H
2
gas
Carbonyls 2,4,DNP Orange/red crystals
produced
Aldehyde Fehlings solution Blue solution to red
precipitate
Aldehyde Tollens Reagent Silver mirror formed
Carboxylic acid Sodium carbonate Effervescence of CO
2
evolved
1
o
2
o
alcohol and
aldehyde
Sodium dichromate and
sulphuric acid
Orange to green colour
change
chloroalkane Warm with silver nitrate Slow formation of white
precipitate of AgCl
Acyl chloride Silver nitrate Vigorous reaction- steamy
fumes of HCl- rapid white
precipitate of AgCl

N Goalby chemrevise.org 22
Measuring the Enthalpy Change for a Reaction Experimentally
Calorimetric method
If the reaction is slow then the exact temperature rise can be
difficult to obtain as cooling occurs simultaneously with the
reaction
To counteract this we take readings at regular time intervals and
extrapolate the temperature curve/line back to the time the
reactants were added together.
We also take the temperature of the reactants for a few minutes
before they are added together to get a better average
temperature. If the two reactants are solutions then the
temperature of both solutions need to be measured before
addition and an average temperature is used.
For a reaction in solution we use the following equation
energy change = mass of solution x heat capacity x temperature change
Q (J) = m (g) x c
p
(J g
-1
K
-1
) x T ( K)
This equation will only give the
energy for the actual quantities
used. Normally this value is
converted into the energy
change per mole of one of the
reactants. (The enthalpy change
of reaction, H)
General method
washes the equipment (cup and pipettes etc) with the solutions to be used
dry the cup after washing
put polystyrene cup in a beaker for insulation and support
Measure out desired volumes of solutions with volumetric pipettes and transfer to
insulated cup
clamp thermometer into place making sure the thermometer bulb is immersed in
solution
measure the initial temperatures of the solution or both solutions if 2 are used. Do this
every minute for 2-3 minutes
At minute 3 transfer second reagent to cup. If a solid reagent is used then add the
solution to the cup first and then add the solid weighed out on a balance.
If using a solid reagent then use ‘before and after’ weighing method
stirs mixture (ensures that all of the solution is at the same temperature)
Record temperature every minute after addition for several minutes
One type of experiment is one in which substances are mixed
in an insulated container and the temperature rise measured.
This could be a solid dissolving or reacting in a
solution or it could be two solutions reacting
together
Errors in this method
• energy transfer from surroundings (usually loss)
• approximation in specific heat capacity of solution. The method assumes all
solutions have the heat capacity of water.
• neglecting the specific heat capacity of the calorimeter- we ignore any energy
absorbed by the apparatus.
• reaction or dissolving may be incomplete or slow.
• Density of solution is taken to be the same as water.
Calorimetric method
Read question carefully. It
may be necessary to
describe:
• Method
• Drawing of graph with
extrapolation
• Description of the
calculation
CORE PRACTICAL 8: To determine the enthalpy change of a reaction using Hess’s Law

Calculating the enthalpy change of reaction, H
r
from experimental data
General method
1. Using q= m x c
p
x T calculate energy change for quantities used
2. Work out the moles of the reactants used
3. Divide q by the number of moles of the reactant not in excess to give H
4. Add a sign and unit (divide by a thousand to convert Jmol
-1
to kJmol
-1
The heat capacity of water is 4.18
J g
-1
K
-1
. In any reaction where the
reactants are dissolved in water
we assume that the heat capacity
is the same as pure water.
Also assume that the solutions
have the density of water, which is
1g cm
-3
. Eg 25cm
3
will weigh 25 g
Example 3. Calculate the enthalpy change of reaction for the reaction where 25.0cm
3
of 0.20M
copper sulphate was reacted with 0.01mol (excess of zinc). The temperature increased 7.0
o
C .
Step 1: Calculate the energy change for the amount of reactants in the test tube.
Q = m x c
p
x T
Q = 25 x 4.18 x 7
Q = 731.5 J
Step 2 : calculate the number of moles of the reactant not in excess.
moles of CuSO
4
= conc x vol
= 0.2 x 25/1000
= 0.005 mol
If you are not told what is in excess, then you need to work
out the moles of both reactants and work out using the
balanced equation which one is in excess.
Step 3 : calculate the enthalpy change per mole which is often called H (the enthalpy change of reaction)
H = Q/ no of moles
= 731.5/0.005
= 146300 J mol
-1
= 146 kJ mol
-1
to 3 sf
Finally add in the sign to represent the energy change: if temp increases the
reaction is exothermic and is given a minus sign e.g. –146 kJ mol
-1
Remember in these
questions: sign, unit
Example 4. 25.0cm
3
of 2.0M HCl was neutralised by 25.0cm
3
of 2.0M NaOH. The Temperature increased 13.5
o
C
What was the energy change per mole of HCl?
Step 1: Calculate the energy change for the amount of reactants in the test tube.
Q = m x c
p
x T
Q = 50 x 4.18 x13.5
Q = 2821.5 J
Step 2 : calculate the number of moles of the HCl.
moles of HCl = conc x vol
= 2 x 25/1000
= 0. 05 mol
Step 3 : calculate H the enthalpy change per mole which might be called the enthalpy change of neutralisation
H = Q/ no of moles
= 2821.5/0.05
= 564300 J mol
-1
= -56.4 kJ mol
-1
to 3 sf
Exothermic and so is given a minus sign
Remember in these
questions: sign, unit,
Note the mass equals the mass of acid + the
mass of alkali, as they are both solutions.
Note the mass is the mass of the copper sulphate
solution only. Do not include mass of zinc powder.
23
N Goalby chemrevise.org

N Goalby chemrevise.org 24
CuSO
4
(aq)
CuSO
4 (s)
+ 5H
2
O
(l)
CuSO
4
.5H
2
O
(s)
+ 11kJmol
-1
= -66.1 kJ mol
-1
H reaction
+ aq
+ aq
H reaction
+11kJ mol
-1
-66.1 kJ mol
-1
H reaction
= -66.1
- 11
= -77.1 kJ mol
-1
This Hess’s law is used to work out the
enthalpy change to form a hydrated salt from
an anhydrous salt.
This cannot be done experimentally because it
is impossible to add the exact amount of water
without the solid dissolving and it is not easy to
measure the temperature change of a solid.
Often Hess’s law cycles are used to measure the enthalpy change for a reaction that cannot be measured directly by
experiments. Instead alternative reactions are carried out that can be measured experimentally.
Instead both salts are dissolved in excess water
to form a solution of copper sulphate. The
temperature changes can be measured for
these reactions.
Detailed method for measuring enthalpy change of solution of anhydrous copper(II) sulfate
1. Weigh out between 3.90 g and 4.10 g of anhydrous copper(II) sulfate in a dry weighing bottle. The precise mass
should be recorded.
2. Using a volumetric pipette, place 25 cm
3
of deionised water into a polystyrene cup and record its temperature at the
beginning (t=0), start the timer and then record the temperature again every minute, stirring the liquid continuously.
3. At the fourth minute, add the powdered anhydrous copper(II) sulfate rapidly to the water in the polystyrene cup and
continue to stir, but do not record the temperature.
4. Reweigh the empty weighing bottle
5. At the fifth minute and for every minute up to 15 minutes, stir and record the temperature of the solution in the
polystyrene cup.
6. Plot a graph of temperature (on the y-axis) against time. Draw two separate best fit lines; one, which joins the points
before the addition, and one, which joins the points after the addition, extrapolating both lines to the fourth minute.
7. Use your graph to determine the temperature change at the fourth minute, which theoretically should have
occurred immediately on addition of the solid.
8. Using q= m x c
p
x T calculate energy change
= 20 x 4.18 x T
9. Calculate H
solution
by dividing q by number of moles of anhydrous copper(II) sulfate in mass added
The above method is then repeated using hydrated copper sulfate. The two H
solution
can then be used to calculate
the H for the enthalpy change of forming a hydrated salt as in the example above
25
Example 5. Calculate the enthalpy change of combustion for the reaction where 0.65g of propan-1-ol was
completely combusted and used to heat up 150g of water from 20.1 to 45.5
o
C
Step 1: Calculate the energy change used to heat up the water.
Q = m x c
p
x T
Q = 150 x 4.18 x 25.4
Q = 15925.8 J
Step 2 : calculate the number of moles of alcohol combusted.
moles of propan-1-ol = mass/ Mr
= 0.65 / 60
= 0.01083 mol
Step 3 : calculate the enthalpy change per mole which is called Hc (the enthalpy change of combustion)
H = Q/ no of moles
= 15925.8/0.01083
= 1470073 J mol
-1
= 1470 kJ mol
-1
to 3 sf
Finally add in the sign to represent the energy change: if temp increases the
reaction is exothermic and is given a minus sign eg –1470 kJ mol
-1
Remember in these
questions: sign, unit
Note the mass in this equation is the mass of water in
the calorimeter and not the alcohol
Enthalpies of combustion can be calculated by using calorimetry.
Generally the fuel is burnt and the flame is used to heat up water in a
metal cup.
Need to measure
• mass of spirit burner before and after
• Temperature change of water
• Volume of water in cup
N Goalby chemrevise.org
Measuring Enthalpies of Combustion using Flame Calorimetry
• energy losses from calorimeter
• Incomplete combustion of fuel
• Incomplete transfer of energy
• Evaporation of fuel after weighing
• Heat capacity of calorimeter not included
• Measurements not carried out under standard conditions as H
2
O is
gas, not liquid, in this experiment
Errors in this method

26
CORE PRACTICAL 9: Finding the Ka value for a weak acid – titration curves
Strong acid – Strong base
pH
7
1
13
25
cm
3
of base
Long steep part from
around 3 to 9
pH at equivalence
point = 7
The equivalence point lies
at the mid point of the
extrapolated vertical
portion of the curve.
e.g. HCl and NaOH
N Goalby chemrevise.org
Weak acid – Strong base
e.g. CH
3
CO
2
H and NaOH
pH
7
1
13
V
cm
3
of base
At the start the pH rises quickly and then levels off. The
flattened part is called the buffer region and is formed because
a buffer solution is made
Steep part of curve >7 (around
7 to 9)
Equivalence point >7
pH starts
near 3
Half neutralisation volume
[H
+
(aq)
][A
-
(aq)
]
[HA (aq)]
Ka=
For weak acids
At ½ the neutralisation volume
the [HA] = [A
-
]
So Ka= [H
+
] and pKa = pH
If we know the Ka we can then work out the pH
at ½ V or vice versa.
If a pH curve is plotted then the pH of a weak
acid at half neutralisation (½ V) will equal the pKa
Constructing a pH curve
Calibrate meter first by measuring known pH of a
buffer solution. This is necessary because pH meters
can lose accuracy on storage.
Most pH probes are calibrated by putting probe in a
set buffer (often pH 4) and pressing a calibration
button/setting for that pH. Sometimes this is
repeated with a second buffer at a different pH
Can also improve accuracy by maintaining
constant temperature
1. Transfer 25cm
3
of acid to a conical flask with a volumetric
pipette
2. Measure initial pH of the acid with a pH meter
3. Add alkali in small amounts (2cm
3
) noting the volume
added
4. Stir mixture to equalise the pH
5. Measure and record the pH to 1 d.p.
6. Repeat steps 3-5 but when approaching endpoint add in
smaller volumes of alkali
7. Add until alkali in excess
½ V
pH
7
1
13
25
cm
3
of base
Strong acid – Weak base
Equivalence point < 7
Steep part of curve <7
(around 4 to 7)
Weak acid – Weak base
e.g. CH
3
CO
2
H and NH
3
e.g. HCl and NH
3
pH
7
1
13
25
cm
3
of base
No Steep part of the curve

27
CORE PRACTICAL 10: Investigating some electrochemical cells
Salt Bridge
The salt bridge is used to connect up the circuit. The free moving ions conduct the charge.
A salt bridge is usually made from a piece of filter paper (or material) soaked in a salt solution, usually Potassium Nitrate.
It can also be a glass U tube containing a salt solution plugged with cotton wool
The salt should be unreactive with the electrodes and electrode solutions.. E.g. potassium chloride would not be suitable
for copper systems because chloride ions can form complexes with copper ions.
A wire is not used because the metal wire would set up its own electrode system with the solutions.
N Goalby chemrevise.org
Zinc
electrode
copper
electrode
1M zinc
sulphate
solution
1M
copper
sulphate
solution
Salt bridge
Electron
flow
Method
• Clean the zinc and copper foils with emery before use.
Degrease the metal using some cotton wool and
propanone.
• Place the copper strip into a 100 cm
3
beaker with
about 50 cm
3
of 1 mol dm
–3
CuSO
4
solution.
• Place the zinc strip into a 100 cm
3
beaker with about
50 cm
3
of 1 mol dm
–3
ZnSO
4
solution.
• Use a strip of filter paper soaked in saturated
potassium nitrate solution for the salt bridge
• Connect the Cu(s)|Cu
2+
(aq) and Zn(s)|Zn
2+
(aq) half-
cells by connecting the metals using the crocodile clips
and leads provided to the voltmeter
Method
If one or both of the half cells do not contain a
conducting metal, we must use an inert platinum
electrode.
Set up a copper half cell using a similar
arrangement to the previous one. Combine it
with a Fe
2+
/Fe
3+
half-cell with a platinum
electrode.
The half cell should have a mixture of acidified
1.0M iron(II) sulphate solution and an equal
volume of 0.5M iron(III) sulphate solution as the
electrolyte. Use a fresh salt bridge.
1M FeSO
4
and 0.5 M Fe
2
(SO
4
)
3
Salt bridge
KNO
3
(aq)
Note: in the electrode system containing two solutions it is
necessary to use a platinum electrode and both ion solutions must
be of a 1M concentration so [Fe
2+
] = 1M and [Fe
3+
] = 1M .
copper
electrode
1M
copper
sulphate
solution
Pt electrode
A platinum electrode is used because it is
unreactive and can conduct electricity

N Goalby chemrevise.org 28
. Detailed Procedure : how much iron in iron tablets
• Weigh accurately two 'ferrous sulphate' tablets.
• Grind up the tablets with a little 1M sulphuric acid, using a pestle and mortar.
• Through a funnel, transfer the resulting paste into a 100cm
3
volumetric flask. Use further small volumes of 1 M
sulphuric acid to rinse the ground-up tablets into the flask.
• Then add sufficient 1M sulphuric acid to make up the solution to exactly 100cm
3
. Stopper the flask and shake it to
make sure that all the contents are thoroughly mixed. They will not all be in solution although the Fe
2+
ions which
were present in the tablets will be dissolved.
• Titrate 10.0 cm
3
portions of the solution with 0.0050 M potassium manganate(VII). The end-point is marked by the
first permanent purple colour.
Manganate Redox Titrations
The redox titration between Fe
2+
with MnO
4
–
(purple) is a very common
exercise. This titration is self indicating because of the significant colour
change from reactant to product.
MnO
4
-
(aq) + 8H
+
(aq) + 5Fe
2+
(aq) Mn
2+
(aq) + 4H
2
O (l) + 5Fe
3+
(aq)
Purple colourless
The purple colour of manganate can make it
difficult to see the bottom of meniscus in
the burette.
If the manganate is in the burette then the end
point of the titration will be the first permanent
pink colour.
Colourless purple
Choosing correct acid for manganate titrations.
The acid is needed to supply the 8H
+
ions. Some acids are not suitable as they set up alternative redox reactions and
hence make the titration readings inaccurate.
Only use dilute sulphuric acid for manganate titrations.
Insufficient volumes of sulphuric acid will mean the solution is not acidic enough and MnO
2
will be produced instead
of Mn
2+
.
MnO
4
-
(aq) + 4H
+
(aq) + 3e- MnO
2
(s) + 2H
2
O
The brown MnO
2
will mask the colour change and lead to a greater (inaccurate) volume of Manganate being used in
the titration.
Using a weak acid like ethanoic acid would have the same effect as it cannot supply the large amount of hydrogen
ions needed (8H
+
).
It cannot be conc HCl as the Cl
-
ions would be oxidised to Cl
2
by MnO
4
-
as the E
o
MnO
4
-
/Mn
2+
> E
o
Cl
2
/Cl
-
MnO
4
-
(aq) + 8H
+
(aq) + 5e
–
Mn
2+
(aq) + 4H
2
O(l) E+1.51V
Cl
2
(aq) +2e
–
2Cl
–
(aq) E +1.36V
This would lead to a greater volume of manganate being used and poisonous Cl
2
being produced.
It cannot be nitric acid as it is an oxidising agent. It oxidises Fe
2+
to Fe
3+
as E
o
NO
3
-
/HNO
2
> E
o
Fe
3+
/Fe
2+
NO
3
-
(aq) + 3H
+
(aq) + 2e
–
HNO
2
(aq) + H
2
O(l) E
o
+0.94V
Fe
3+
(aq)+e
–
Fe
2+
(aq) E
o
+0.77 V
This would lead to a smaller volume of manganate being used.
A 2.41g nail made from an alloy containing iron is
dissolved in 100cm
3
acid. The solution formed contains
Fe(II) ions.
10cm
3
portions of this solution are titrated with
potassium manganate (VII) solution of 0.02M. 9.80cm
3
of KMnO
4
were needed to react with the solution
containing the iron.
What is the percentage of Iron by mass in the nail?
Example 6 Manganate titration
MnO
4
-
(aq)
+ 8H
+
(aq)
+ 5Fe
2+
Mn
2+
(aq)
+ 4H
2
O + 5Fe
3+
Step1 : find moles of KMnO
4
moles = conc x vol
0.02 x 9.8/1000
= 1.96x10
-4
mol
Step 2 : using balanced equation find moles Fe
2+
in 10cm
3
= moles of KMnO
4
x 5
= 9.8x10
-4
mol
Step 3 : find moles Fe
2+
in 100cm
3
= 9.8x10
-4
mol x 10
= 9.8x10
-3
mol
Step 4 : find mass of Fe in 9.8x10
-3
mol
mass= moles x RAM = 9.8x10
-3
x 55.8 = 0.547g
Step 5 : find % mass
%mass = 0.547/2.41 x100
= 22.6%
CORE PRACTICAL 11: Redox titration

29
Other useful manganate titrations
With hydrogen peroxide
Ox H
2
O
2
O
2
+ 2H
+
+ 2e
-
Red MnO
4
-
(aq) + 8H
+
(aq) + 5e
-
Mn
2+
(aq) + 4H
2
O
Overall 2MnO
4
-
(aq) + 6H
+
(aq) + 5H
2
O
2
5O
2
+ 2Mn
2+
(aq) + 8H
2
O
Ox C
2
O
4
2-
2CO
2
+ 2e
-
Red MnO
4
-
(aq) + 8H
+
(aq) + 5e
-
Mn
2+
(aq) + 4H
2
O
Overall 2MnO
4
-
(aq) + 16H
+
(aq) + 5C
2
O
4
2-
(aq) 10CO
2
(g) + 2Mn
2+
(aq) + 8H
2
O(l)
With ethanedioate
With Iron (II) ethanedioate both the Fe
2+
and the C
2
O
4
2-
react with the MnO
4
-
1MnO
4
-
reacts with 5Fe
2+
and 2 MnO
4
-
reacts with 5C
2
O
4
2-
MnO
4
-
(aq) + 8H
+
(aq) + 5Fe
2+
Mn
2+
(aq) + 4H
2
O + 5Fe
3+
2MnO
4
-
(aq) + 16H
+
(aq) + 5C
2
O
4
2-
10CO
2
+ 2Mn
2+
(aq) + 8H
2
O
So overall
3MnO
4
-
(aq) + 24H
+
(aq) + 5FeC
2
O
4
10CO
2
+ 3Mn
2+
(aq) + 5Fe
3+
+ 12H
2
O
So overall the ratio is 3 MnO
4
-
to 5 FeC
2
O
4
The reaction between MnO
4
-
and
C
2
O
4
2-
is slow to begin with (as the
reaction is between two negative
ions). To do as a titration the conical
flask can be heated to 60
o
C to
speed up the initial reaction.
N Goalby chemrevise.org
Example 7
A 1.412 g sample of impure FeC
2
O
4
.2H
2
O was
dissolved in an excess of dilute sulphuric acid
and made up to 250 cm
3
of solution. 25.0 cm
3
of this solution decolourised 23.45 cm
3
of a
0.0189 mol dm
–3
solution of potassium
manganate(VII).
What is the percentage by mass of
FeC
2
O
4
.2H
2
O in the original sample?
Step1 : find moles of KMnO
4
moles = conc x vol
0.0189 x 23.45/1000
= 4.43x10
-4
mol
Step 2 : using balanced equation find moles FeC
2
O
4
.2H
2
O in 25cm
3
= moles of KMnO
4
x 5/3 (see above for ratio)
= 7.39x10
-4
mol
Step 3 : find moles FeC
2
O
4
.2H
2
O in 250 cm
3
= 7.39x10
-4
mol x 10
= 7.39x10
-3
mol
Step 4 : find mass of FeC
2
O
4
.2H
2
O in 7.39x10
-3
mol
mass= moles x Mr = 7.39x10
-3
x 179.8 = 1.33g
Step 5 ; find % mass
%mass = 1.33/1.412 x100
= 94.1%
EDTA titrations
The formation of the stable EDTA complex with metal ions can with the choice of suitable indicator be done in a
quantitative titration.
[Cu(H
2
O)
6
]
2+
+ EDTA
4-
[Cu(EDTA)]
2-
+ 6H
2
O
Always the same 1:1 ratio with any metal ion
Example 8
A river was polluted with copper(II)
ions. 25.0 cm
3
sample of the river
water was titrated with a 0.0150
mol dm
–3
solution of EDTA
4–
, 6.45
cm
3
were required for complete
reaction.
Calculate the concentration, in mol
dm
–3
, of copper(II) ions in the river
water.
Step1 : find moles of EDTA
4-
moles = conc x vol = 0.0150 x 6.45/1000
= 9.68x10
-5
mol
Step 2 : using balanced equation find moles Cu
2+
1:1 ratio
= 9.68x10
-5
mol
Step 3 : find conc Cu
2+
in 25cm
3
= 9.68x10
-5
/0.025
= 0.00387 moldm
-3

N Goalby chemrevise.org 30
1. Add 1.5 g of hydrated copper(II) sulfate to weighing
bottle and measure the combined mass.
2. Transfer the copper (II) sulfate to a test tube and
measure the mass of the empty weighing bottle.
3. Add 4 cm
3
of water to the test tube
4. Place the test tube in the water bath (a beaker with
freshly boiled water).
5. Stir gently to dissolve the copper(II) sulfate.
6. Remove the test tube containing copper(II) sulfate
solution from the water bath
7. In a fume cupboard, add 2 cm
3
of concentrated
ammonia solution to the copper(II) sulfate solution
8. Pour the contents of the test tube into a beaker
containing 6 cm
3
of ethanol. Stir well and cool the
mixture in an ice bath.
9. Using a Buchner funnel and flask, filter the crystals.
Wash the test tube with some cold ethanol and add
the washings to the Buchner funnel. Finally, rinse the
crystals with a little cold ethanol.
10. Scrape the crystals onto a fresh piece of filter paper
and cover with a second piece of filter paper and
press to dry the crystals.
11. Measure the mass of the dry crystals
The mass of the copper(II) sulfate is the
difference between the two masses.
Buchner flask (has
thicker glass walls than
a normal flask to cope
with the vacuum )
Filter paper
This is vacuum filtration. The apparatus is
connected to a water pump which will
produce a vacuum. Use if larger amounts of
solid are formed.
Air outlet to
water pump
Buchner
funnel
Concentrated ammonia is corrosive.
Use in a fume cupboard and wear
gloves
Core practical 12: Prepare a transition metal complex
Equation for reaction
CuSO
4
•5H
2
O + 4NH
3
→ Cu(NH
3
)
4
SO
4
•H
2
O + 4H
2
O
Loss of yield in this process
• Crystals lost when filtering or washing
• Some product stays in solution after recrystallisation
• other side reactions occurring
If the crystals are not dried properly the mass will be larger
than expected which can lead to a percentage yield >100%

N Goalby chemrevise.org
31
Techniques to investigate rates of reaction
measurement of the change in volume of a gas
Titrating samples of reaction mixture with acid, alkali, sodium thiosulphate etc
Colorimetry.
Measurement of optical activity.
Measurement of change of mass
Measuring change in electrical conductivity
H
2
O
2
(aq) + 2I
-
(aq) + 2H
+
(aq) 2H
2
O(l) + I
2
(aq)
HCOOCH
3
(aq) + NaOH(aq) HCOONa(aq) + CH
3
OH(aq)
(CH
3
)
2
C=CH
2
(g) + HI(g) (CH
3
)
3
CI(g)
BrO
3
–
(aq) + 5Br
–
(aq) + 6H
+
(aq) 3Br
2
(aq) + 3H
2
O(l)
HCOOH(aq) + Br
2
(aq) 2H
+
(aq) + 2Br
-
(aq) + CO
2
(g)
HCOOH(aq) + Br
2
(aq) 2H
+
(aq) + 2Br
-
(aq) + CO
2
(g)
There are several different methods for measuring reactions rates. Some reactions can be measured
in several ways
This works if there is a change in the number of moles of
gas in the reaction. Using a gas syringe is a common
way of following this.
This works if there is a gas produced which is allowed to
escape. Works better with heavy gases such as CO
2
HCOOH(aq) + Br
2
(aq) 2H
+
(aq) + 2Br
-
(aq) + CO
2
(g)
CH
3
COCH
3
(aq) + I
2
(aq) → CH
3
COCH
2
I(aq) + H
+
(aq) + I
–
(aq)
Small samples are removed from the reaction mixture, quenched (which
stops the reaction) and the titrated with a suitable reagent.
The NaOH could be titrated with an acid
The H
+
could be titrated with an alkali
The I
2
could be titrated with sodium
thiosulphate
If one of the reactants or products is coloured
then colorimetry can be used to measure the
change in colour of the reacting mixtures
The I
2
produced is a brown solution
Can be used if there is a change in the number
of ions in the reaction mixture
If there is a change in the optical activity through
the reaction this could be followed in a
polarimeter
CH
3
CHBrCH
3
(l) + OH
−
(aq) CH
3
CH(OH)CH
3
(l) + Br
−
(aq)
Investigating rates of reaction

32N Goalby chemrevise.org
When we follow one experiment over time recording the change in
concentration it is the continuous rate method.
The gradient represents the rate of reaction. The reaction is
fastest at the start where the gradient is steepest. The rate
drops as the reactants start to get used up and their
concentration drops. The graph will eventual become
horizontal and the gradient becomes zero which represents
the reaction having stopped.
time
concentration
Measurement of the change in volume of a gas
Mg + HCl MgCl
2
+H
2
This works if there is a change in the number of moles of gas
in the reaction. Using a gas syringe is a common way of
following this. It works quite well for measuring continuous
rate but a typical gas syringe only measures 100ml of gas so
you don’t want at reaction to produce more than this volume.
Quantities of reactants need to be calculated carefully.
gas syringe
Typical Method
• Measure 50 cm
3
of the 1.0 mol dm
–3
hydrochloric acid
and add to conical flask.
• Set up the gas syringe in the stand
• Weigh 0.20 g of magnesium.
• Add the magnesium ribbon to the conical flask, place the
bung firmly into the top of the flask and start the timer.
• Record the volume of hydrogen gas collected every 15
seconds for 3 minutes.
The initial rate is the rate at the start of the
reaction, where it is fastest. It is obtained by taking
the gradient of a continuous monitoring conc vs
time graph at time = zero. A measure of initial rate
is preferable as we know the concentrations at the
start of the reaction
Large Excess of reactants
In reactions where there are several reactants, if the
concentration of one of the reactant is kept in a large excess
then that reactant will appear not to affect rate and will be
pseudo-zero order . This is because its concentration stays
virtually constant and does not affect rate.
General: Measuring the rate of reaction: by an continuous monitoring method
Continuous rate data
This is data from one experiment where the concentration of a
substance is followed throughout the experiment.
Continuous rate experiments
This data is processed by plotting the data and calculating
successive half-lives.
If half-lives are constant
then the order is 1
st
order
The half-life of a first-order reaction
is independent of the concentration and is constant
If half-lives rapidly increase then the order is
2nd order
0.060
0.030
0.015
0.0075
t ½
t ½
t ½
Time (min)
[A]

Propanone reacts with iodine in acidic solution (the acid is a catalyst) as shown in the equation
below.
CH
3
COCH
3
(aq) + I
2
(aq) → CH
3
COCH
2
I(aq) + H
+
(aq) + I
–
(aq)
The rate equation for the reaction is
Rate = k[CH
3
COCH
3
(aq)][H
+
(aq)]
This reaction can be followed by removing small samples from the reaction mixture with a
volumetric pipette. The sample is then quenched by adding excess sodium hydrogencarbonate to
neutralize acid catalyst which stops the reaction. Then the sample can be titrated with sodium
thiosulphate using a starch catalyst
2S
2
O
3
2-
(aq) + I
2
(aq) 2I
-
(aq) + S
4
O
6
2-
(aq)
yellow/brown sol colourless sol
Time (min)
[I
2
]
This reaction is zero order with respect to I
2
but 1
st
order
with respect to the propanone and acid catalyst
If there is a zero order reactant there must
be at least two steps in the mechanism
because the rate determining step will not
involve the zero order reactant
The rate determining step of this reaction must therefore contain one propanone molecule and one H
+
ion
forming an intermediate. The iodine will be involved in a subsequent faster step.
Detailed Method
1. To a beaker add 25 cm
3
of 1 mol dm
−3
aqueous propanone and 25 cm
3
of 1 mol dm
−3
sulfuric acid.
2. Add 50 cm
3
of 0.02 mol dm
− 3
iodine solution. Start the clock swirl the beaker well to mix.
3. Using a 10cm
3
pipette, withdraw a sample of the mixture and transfer it to a conical flask.
4. Add a spatula measure of sodium hydrogencarbonate (This stops the reaction). Record the time at which the sodium
hydrogencarbonate is added.
5. Titrate the iodine present in the conical flask with 0.01 mol dm
− 3
sodium thiosulfate solution. When the colour
turns a pale yellow add the starch indicator. The end point is then when the mixture goes from blue to colourless.
6 Every 5 minutes withdraw another 10 cm
3
sample and repeat steps 4 and 5
33N Goalby chemrevise.org
This method allows the order with respect to Iodine to be calculated because the propanone and
acid are in large excess so their concentrations do not change during the reaction
CORE PRACTICAL 13a: Rates of reaction: Following the rate of the iodine propanone
reaction by a titrimetric method

N Goalby chemrevise.org
34
Detailed method
• Put each of the chemicals in the table in separate burettes.
•
In each experiment, measure out required volumes of the potassium iodide, sodium thiosulphate, starch and
water into a small conical flask from the burettes
• Measure the hydrogen peroxide into a test tube
• Pour the hydrogen peroxide from the test tube into the conical flaks and immediately start the timer. Stir the
mixture.
• Time until the first hint of blue/ black colour appears
CORE PRACTICAL 13b: Rates of reaction: clock Reaction
The initial rate can be calculated from taking the
gradient of a continuous monitoring conc vs time
graph at time = zero
Initial rate can also be calculated from clock reactions
where the time taken to reach a fixed concentration is
measured.
A Common Clock Reaction (no need to learn details)
Hydrogen peroxide reacts with iodide ions to form iodine. The thiosulfate
ion then immediately reacts with iodine formed in the second reaction as
shown below.
H
2
O
2
(aq) + 2H
+
(aq) + 2I
–
(aq) → I
2
(aq) + 2H
2
O(l)
2S
2
O
3
2–
(aq) + I
2
(aq) → 2I
–
(aq) + S
4
O
6
2–
(aq)
When the I
2
produced has reacted with all of the limited amount of
thiosulfate ions present, excess I
2
remains in solution. Reaction with the
starch then suddenly forms a dark blue-black colour.
A series of experiments is carried out, in which the concentration of iodide
ions is varied, while keeping the concentrations of all of the other reagents
the same. In each experiment the time taken (t) for the reaction mixture to
turn blue is measured.
In clock reactions there are often two
successive reactions and an end point is
achieved when one limited reactant runs
out, resulting in a sudden colour change
By repeating the experiment several times,
varying the concentration of a reactant e.g.
I
–
, ( keeping the other reactants at constant
concentration )you can determine the
order of reaction with respect to that
reactant
The initial rate of the reaction can be
represented as (1/t )
Experiment
Sulfuric
acid (H
+
) ml Starch ml Water ml
Potassium
iodide(I
-
) ml
Sodium
Thiosulfate S
2
O
3
2-
ml
1 25 1 20 5 5
2 25 1 15 10 5
3 25 1 10 15 5
4 25 1 5 20 5
5 25 1 0 25 5
Hydrogen
peroxide ml
10
10
10
10
10
Working out rate order graphically
In an experiment where the concentration of one of
the reagents is changed and the reaction rate measured
it is possible to calculate the order graphically
Rate = k [I
-
]
n
Log both sides of equation
Log rate = log k + n log [Y]
A graph of log rate vs log [I
-
] will yield a straight line where the
gradient is equal to the order n
Taking rate equation
Y = c + m x
In this experiment high concentrations with quick
times will have the biggest percentage errors.
Normally to work out the rate equation we do a series of experiments where the initial concentrations of reactants are
changed (one at a time) and measure the initial rate each time.
log (rate)
log [I
-
]
y intercept = log K
gradient = n
= change in y
change in x

N Goalby chemrevise.org
35
Experiment Determine the activation energy for the reaction between bromide ions and bromate(V) ions
The Arrhenius equation can be rearranged
ln k = constant – Ea/(RT)
k is proportional to the rate of reaction so ln k can be
replaced by ln(rate)
From plotting a graph of ln(rate) or ln k against 1/T the
activation energy can be calculated from measuring the
gradient of the line
ln (Rate)
1/T
Gradient = - Ea/ R
Ea = - gradient x R
-4.1
-3.6
-3.1
-2.6
-2.1
-1.6
0.0029 0.003 0.0031 0.0032 0.0033 0.0034
ln (Rate)
1/T
use a line of best fit
use all graph paper
choose points far apart on the graph to
calculate the gradient
Temperature
T (K) 1/T
time t
(s) 1/t Ln (1/t)
297.3 0.003364 53 0.018868 -3.9703
310.6 0.00322 24 0.041667 -3.1781
317.2 0.003153 16 0.0625 -2.7726
323.9 0.003087 12 0.083333 -2.4849
335.6 0.00298 6 0.166667 -1.7918
x
1
,y
1
x
2
,y
2
gradient = y
2
-y
1
x
2
-x
1
In above example gradient =-5680
The gradient should
always be -ve
Ea = - gradient x R (8.31)
= - -5680 x8.31
= 47200 J mol
-1
The unit of Ea using this equation will be J mol
-1
. Convert
into kJ mol
-1
by dividing 1000
Ea = +47.2 kJ mol
-1
Example 9
Analysis of results to calculate Activation Energy
CORE PRACTICAL 14: Finding the activation energy of a reaction
Detailed Method
1. Pipette 10 cm
3
of phenol solution and 10 cm
3
of bromide/bromate solution into a boiling tube.
2. Add a few drops of methyl red indicator to the mixture.
3. Into a second boiling tube pipette 5 cm
3
of sulfuric acid.
4. Place the two boiling tubes in a water bath at temperature 20
o
C.
5. When the substances have reached the water temperature, mix the contents into one of the boiling tubes and
swirl. Start the stop clock.
6. Place the boiling tube containing the reaction mixture in the water bath.
7. Stop the clock when the methyl red indicator disappears.
8. Repeat the experiment at 30,40 50 60
o
C
In this experiment rate is 1/time where the time is the time taken for the indicator to change colour.
This is an approximation for initial rate of reaction as it does not include the change in concentration term. We can use
this because we can assume the amount of phenol used in each experiment is the same and constant. The change in
concentration is therefore the same for each experiment so only the time taken to reach this concentration is relevant.
C
6
H
5
OH + 3Br
2
→ C
6
H
2
Br
3
OH + 3HBr
BrO
3
–
+ 5Br
–
+ 6H
+
3Br
2
+ 3H
2
O
The Bromine produced in the first reaction reacts with the phenol. When
the phenol is used up, the bromine is no longer removed. The bromine
then bleaches the methyl red indicator at the ‘end of the reaction’

36
Step Reason
1. Dissolve the impure compound in a minimum volume
of hot (near boiling) solvent.
An appropriate solvent is one which will dissolve both
compound and impurities when hot and one in which the
compound itself does not dissolve well when cold.
The minimum volume is used to obtain saturated
solution and to enable crystallisation on cooling
2. Hot filter solution through (fluted) filter paper quickly. This step will remove any insoluble impurities and heat
will prevent crystals reforming during filtration
3. Cool the filtered solution by inserting beaker in ice Crystals will reform but soluble impurities will remain in
solution form because they are present in small quantities
so solution is not saturated. Ice will increase the yield of
crystals
4. Suction filtrate with a Buchner flask to separate out
crystals
The water pump connected to the Buchner flask reduces
the pressure and speeds up the filtration.
5 Wash the crystals with distilled water To remove soluble impurities
6. Dry the crystals between absorbent paper
Purifying an organic solid: Recrystallisation
Loss of yield in this process
• Crystals lost when filtering or washing
• Some product stays in solution after recrystallisation
• other side reactions occurring
buchner flask
Used for purifying aspirin
N Goalby chemrevise.org
If the crystals are not dried properly the mass will be larger
than expected which can lead to a percentage yield >100%
Preparation of a pure organic solid and test of its purity

N Goalby chemrevise.org 37
16. Core activity: The preparation of aspirin
Add to a 50 cm
3
pear-shaped flask 2.0 g of 2-hydroxybenzoic acid and
4 cm
3
of ethanoic anhydride.
To this mixture add 5 drops of 85% phosphoric(v) acid and swirl to
mix, Fit the flask with a reflux condenser and heat the mixture on a
boiling water bath for about 5 minutes. Without cooling the mixture,
carefully add 2 cm
3
of water in one portion down the condenser.
When the vigorous reaction has ended, pour the mixture into 40 cm
3
of cold water in a 100 cm
3
beaker, stir and rub the sides of the beaker
with a stirring rod necessary to induce crystallisation and, finally,
allow the mixture to stand in ice bath to complete crystallisation.
Collect the product by suction filtration and wash it with a little water.
Purification stage: recrystallisation
Using a measuring cylinder, measure out 15 cm
3
of ethanol into a
boiling tube.
Prepare a beaker half-filled with hot water from a kettle at a
temperature of approximately 75 °C.
Use a spatula to add the crude aspirin to the boiling tube with ethanol
and place the tube in the beaker of hot water.
Stir the contents of the boiling tube until all of the aspirin dissolves
into the ethanol.
Pour the hot solution containing dissolved aspirin through a warmed
filter funnel and fluted filter paper to hot filter
Then pour filtrate into 40 cm
3
of water in a conical flask.
Allow the conical flask to cool slowly and white needles of aspirin
should separate.
Cool the whole mixture in an ice bath.
Filter off the purified solid under reduced pressure and allow it to dry
on filter paper.
Record the mass of the dry purified solid
Detailed method for Nitration Procedure
Measure 2.5 cm
3
of methyl benzoate into a small conical flask and then
dissolve it in 5 cm
3
of concentrated sulfuric acid. When the liquid has
dissolved, cool the mixture in ice.
Prepare the nitrating mixture by adding drop by drop 2 cm
3
of
concentrated sulfuric acid to 2 cm
3
of concentrated nitric acid. Cool this
mixture in ice as well.
Now add the nitrating mixture drop by drop from a dropping pipette to
the solution of methyl benzoate. Stir the mixture with a thermometer and
keep the temperature below 10 °C. When the addition is complete, allow
the mixture to stand at room temperature for another 15 minutes.
After this time, pour the reaction mixture on to about 25 g of crushed ice
and stir until all the ice has melted and crystalline methyl 3-nitrobenzoate
has formed.
Then use same purification method as in aspirin above
Conc acids are corrosive- wear
gloves
The acids react together to make
the NO
2
+
ion
This reaction is exothermic so acids
are kept cool and acid is added
dropwise
The excess ethanoic anhydride will
hydrolyse and the contents of the flask
will boil.
The temperature is kept low at this
stage to prevent multiple
substitution of nitro groups on the
benzene ring
Avoid naked flames due to
flammability of ethanol
This step will remove any insoluble
impurities and heat will prevent crystals
reforming during filtration
Soluble impurities will remain in solution form
because they are present in small quantities
so solution is not saturated. Ice will increase
the yield of crystals
CO
2
H
OH
CH
3
C
O
O
CCH
3
O
CO
2
H
O C
O
CH
3
+ CH
3
CO
2
H
+
Aspirin is made from 2-hydroxybenzoic
acid which contains a phenol group. In the
reaction the phenol group is turned into an
ester by reacting it with the reactive
ethanoic anhydride
Ethanoic anhydride is used instead of
acid chlorides because it is cheaper, less
corrosive, less vulnerable to hydrolysis,
and less dangerous to use.
Aspirin
Detailed method for Preparation of Aspirin
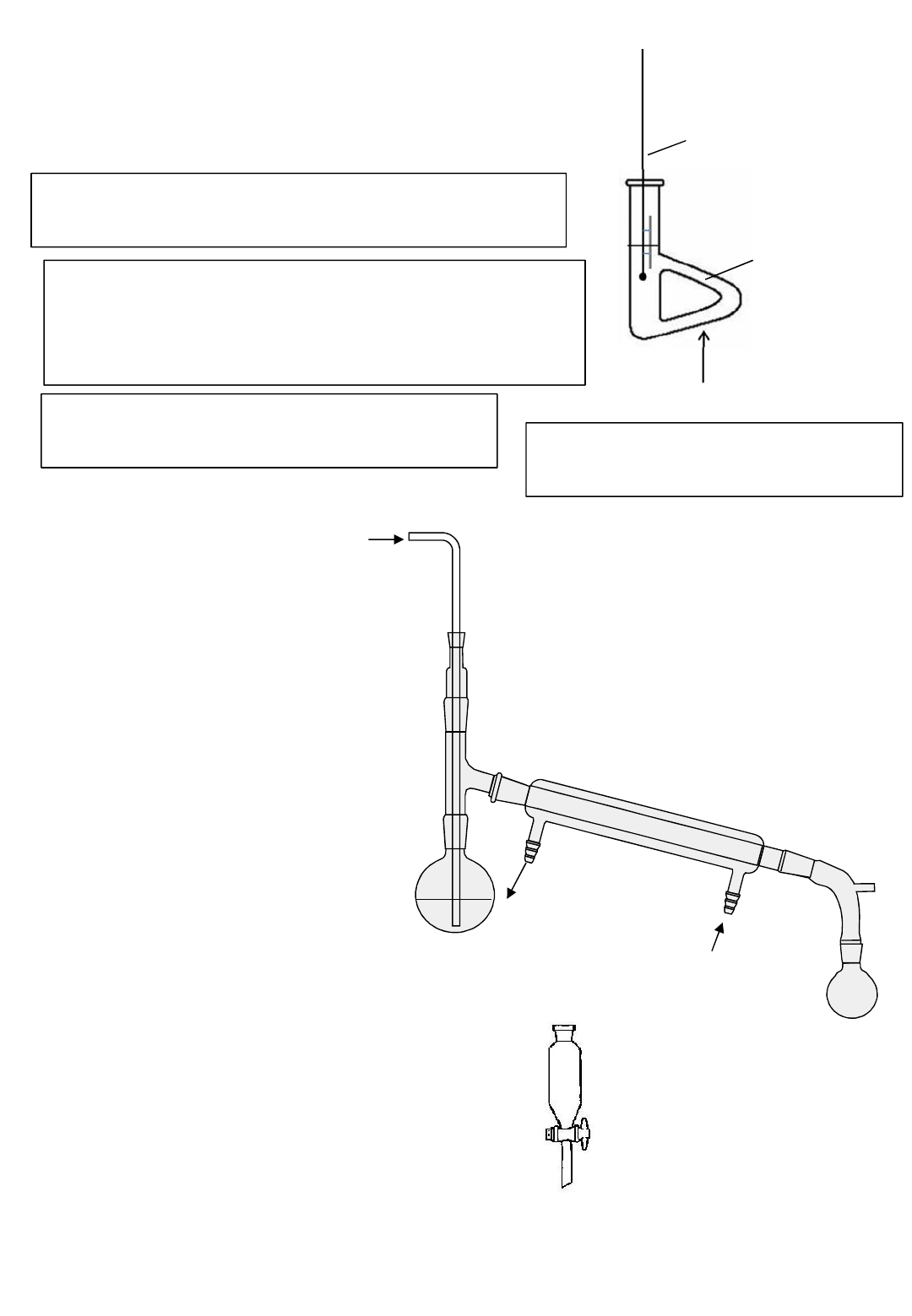
38
N Goalby chemrevise.org
If the sample is very pure then the melting point will be a
sharp one, at the same value as quoted in data books.
One way of testing for the degree of purity is to determine the melting
“point”, or melting range, of the sample.
Heat
Heating oil- needs to
have boiling point
higher than samples
melting point and low
flammability
Thermometer with
capillary tube
strapped to it
containing sample
Measuring melting point
Comparing an experimentally determined melting point value
with one quoted in a data source will verify the degree of
purity.
Sometimes an error may occur if the temperature
on the thermometer is not the same as the
temperature in the actual sample tube.
Melting point can be measured in an electronic melting point machine or
by using a practical set up where the capillary tube is strapped to a
thermometer immersed in some heating oil.
In both cases a small amount of the sample is put into a capillary tube. The
tube is heated up and is heated slowly near the melting point
If impurities are present (and this can include solvent from the
recrystallisation process) the melting point will be lowered and the
sample will melt over a range of several degrees Celsius
In steam distillation steam is passed into
the mixture and the product vapour is
distilled off with the water and condensed
Steam distillation
Water
in
Water
out
steam
in
Advantage of steam distillation:
The product distils at a lower temperature
which can prevents decomposition of the
product if it has a high boiling point
Solvent extraction
Mix organic solvent and oil-water mixture in a
separating funnel then separate the oil layer.
Distil to separate oil from organic solvent
Add anhydrous CaCl
2
to clove oil to dry oil
Decant to remove CaCl
2
Separating funnel

N Goalby chemrevise.org 39
Separation of species by thin-layer chromatography
Method: Thin-layer chromatography
a) Wearing gloves, draw a pencil line 1 cm above the bottom of a
TLC plate and mark spots for each sample, equally spaced along
line.
b) Use a capillary tube to add a tiny drop of each solution to a
different spot and allow the plate to air dry.
c) Add solvent to a chamber or large beaker with a lid so that is no
more than 1cm in depth
d) Place the TLC plate into the chamber, making sure that the level
of the solvent is below the pencil line. Replace the lid to get a
tight seal.
e) When the level of the solvent reaches about 1 cm from the top
of the plate, remove the plate and mark the solvent level with a
pencil. Allow the plate to dry in the fume cupboard.
f) Place the plate under a UV lamp in order to see the spots. Draw
around them lightly in pencil.
g) Calculate the Rf values of the observed spots.
R
f
value = distance moved by amino acid
distance moved by the solvent
Wear plastic gloves to prevent contamination
from the hands to the plate
pencil line –will not dissolve in the solvent
tiny drop – too big a drop will cause different
spots to merge
Depth of solvent– if the solvent is too deep it will
dissolve the sample spots from the plate
Will get more accurate results if the solvent is
allowed to rise to near the top of the plate but the
Rf value can be calculated if the solvent front does
not reach the top of the plate
lid– to prevent evaporation of toxic solvent
dry in a fume cupboard as the solvent is toxic
UV lamp used if the spots are colourless and not
visible
A solid stationary phase separates by adsorption,
A liquid stationary phase separates by relative solubility
If the stationary phase was polar and the moving
phase was non- polar e.g. Hexane. Then non-polar
compounds would pass through the plate more quickly
than polar compounds as they would have a greater
solubility in the non-polar moving phase.
(Think about intermolecular forces)
Separation by chromatography depends on the
balance between solubility in the moving phase and
retention in the stationary phase.
Rf values are used to identify different substances.

N Goalby chemrevise.org
40
Method
Part 1 Preparing the equilibrium mixture
1 Use burettes to prepare a mixture in boiling tube of carboxylic acid, alcohol, and dilute sulfuric acid.
2 Swirl and bung tube. Leave the mixture to reach equilibrium for one week
Part 2 Titrating the equilibrium mixture
1 Rinse a 250 cm
3
volumetric flask with distilled water.
Use a funnel to transfer the contents of the boiling tube into the flask. Rinse the boiling tube with water and add
the washings to the volumetric flask.
2 Use distilled water to make up the solution in the volumetric flask to exactly 250 cm
3
.
Stopper the flask, then invert and shake the contents thoroughly.
3 Use the pipette to transfer 25.0 cm
3
of the diluted equilibrium mixture to a 250 cm
3
conical flask.
4 Add 3 or 4 drops of phenolphthalein indicator to the conical flask.
5 Set up the burette with sodium hydroxide solution..
6 Add the sodium hydroxide solution from the burette until the mixture in the conical flask just turns pink. Record
this burette reading in your table.
7 Repeat the titration until you obtain a minimum of two concordant titres.
Practical: Working out equilibrium constant Kc
The sodium hydroxide will react with the sulphuric acid catalyst and any unreacted carboxylic acid in the
equilibrium mixture
CH
3
CO
2
H + CH
3
CH
2
OH CH
3
CO
2
CH
2
CH
3
+ H
2
O
Ethanoic acid Ethanol Ethyl Ethanoate
A common experiment is working out the equilibrium constant for an esterification reaction. Ethanol and
ethanoic acid are mixed together with a sulphuric acid catalyst.
The pink colour of the phenolphthalein in the titration can fade after the end-point of the titration has been reached
because the addition of sodium hydroxide may make the equilibrium shift towards the reactants
There are many different calculations that can be based on this experiment. Let’s
look at general stages. Not all calculations will use all the stages.
Working out initial amount of moles of reactants
The amount of moles of alcohol and carboxylic acid can be calculated from the densities
and volumes of liquids added
Mass = density x volume
then
Moles = mass X Mr
The initial amount of moles of acid catalyst used is usually determined by titrating a
separate sample of catalyst with sodium hydroxide
Working out equilibrium amount of moles of acid present from the titre results
39.0 cm
3
of 0.400 mol dm
-3
sodium hydroxide was used in the above titration. The initial moles of sulphuric acid
was 5x10
-4
mol. Calculate the moles of ethanoic acid present at equilibrium
Amount of NaOH = vol X conc
= 0.039 x 0.400
= 0.0156 mol
So total amount of H
+
present in 25cm
3
= 0.0156 mol
So total amount of H
+
present in 250cm
3
= 0.156 mol
Total mol acid present = moles of carboxylic acid + moles of acid catalyst
So
Amount of carboxylic acid at equilibrium = 0.156 – (5x10
-4
x 2)
= 0.155 mol
X 2 because H
2
SO
4
has 2 H
+

N Goalby chemrevise.org 41
Calculating the equilibrium constant
Finally calculate the equilibrium constant.
To work out equilibrium concentrations divide the equilibrium amounts by the total volume. Then put in Kc
expression
Kc = [CH
3
CO
2
CH
2
CH
3
] [H
2
O]
[CH
3
CO
2
H] [CH
3
CH
2
OH]
In order to confirm that one week was sufficient time for equilibrium to be established in the mixture
from Part 1, several mixtures could be made and left for different amount of time. If the resulting Kc is
the same value then it can be concluded the time is sufficient
Absorption of visible light is used in spectrometry to
determine the concentration of coloured ions.
method
•Add an appropriate ligand to intensify colour
•Make up solutions of known concentration
•Measure absorption or transmission
•Plot graph of absorption vs concentration
•Measure absorption of unknown and compare
If visible light of increasing frequency is passed through a
sample of a coloured complex ion, some of the light is
absorbed.
The amount of light absorbed is proportional to the
concentration of the absorbing species (and to the distance
travelled through the solution).
Some complexes have only pale colours and do not absorb
light strongly. In these cases a suitable ligand is added to
intensify the colour.
Spectrophotometry
Spectrometers contain a coloured filter. The colour of the filter is chosen to only allow the wavelengths of light
through that would be most strongly absorbed by the coloured solution.
Detailed method- measuring absorption of copper solutions
• Take nine 100cm
3
graduated flasks and pipette 20cm
3
of 2M ammonia solution into each one.
• Use the 0.05M solution of aqueous copper sulphate to make up solutions which are 0.005 to 0.04M
[Cu(NH
3
)
6
]
2+
• Mix each solution thoroughly.
• Insert the red filter into the colorimeter.
• Use a cuvette with distilled water to zero the colorimeter
• Then put each prepared solution in cuvette and measure the absorbance of each solution.
• Plot graph of absorption vs concentration
• Measure absorption of unknown solution and determine its concentration from the calibration curve
Working out equilibrium amount of moles of other substances
Calculate the equilibrium amount of ethanol, ethyl ethanoate and water if there were initially 0.400 mol of
ethanol and 0.500 mol of ethanoic acid and at equilibrium there were 0.155 mol of ethanoic acid.
Amount of ethanoic acid that reacted = initial amount – equilibrium amount
= 0.5 – 0.155
= 0.344mol
Amount of ethanol at equilibrium = initial amount - amount that reacted
= 0.400 – 0.344
= 0.056 mol
Amount of ethyl ethanoate at equilibrium = initial amount + amount that formed
= 0 + 0.344
= 0.344 mol
Amount of water at equilibrium = initial amount + amount that formed
= 0 + 0.344
= 0.344 mol
The amount of water at
equilibrium would not really be
0 as there would be water
present in the acid catalyst

N Goalby chemrevise.org 42
Potassium manganate(VII) will oxidise ethanedioic acid (oxalic acid) to carbon
dioxide and water, in the presence of an excess of acid:
2MnO
4
-
(aq) + 6H
+
(aq) + 5(CO
2
H)
2
(aq) 2Mn
2+
(aq) +10CO
2
(g) + 8H
2
O (I)
Detailed method
1. Prepare a reaction mixture according to the table, using
measuring cylinders.
2. Some members of your group should use Mixture 1 and some
Mixture 2. The results should then be shared.
3. Add 50 cm
3
of 0.02 M potassium manganate(VII) and start timing.
Shake the mixture for about half a minute to mix it well.
4. After about a minute use a pipette to withdraw a 10.0 cm
3
portion of the reaction mixture and empty into a conical flask.
5. Note the time and add about 10cm
3
of 0.1 M potassium iodide
solution. This stops the reaction and releases iodine equivalent to
the remaining manganate.(VII) ions.
6. Titrate the liberated iodine with 0.01 M sodium thiosulphate,
adding a little starch solution near the end-point Record the titre
of sodium thiosulphate.
7. Remove further portions every 3 or 4 minutes and titrate them in
the same way. Continue until the titre is less than 3 cm
3
,
Solution
Mixture 1
Mixture 2
0.2 M ethanedioic acid
100cm
3
100cm
3
0.2 M manganese(ll)
sulphate
-- 15cm
3
1 M sulphuric acid
10 cm
3
10cm
3
Water
90 cm
3
75 cm
3
The autocatalysis by Mn
2+
in titrations of C
2
O
4
2-
with MnO
4
-
overall 2 MnO
4
-
+ 5 C
2
O
4
2-
+ 16 H
+
2Mn
2+
+ 10 CO
2
+ 8 H
2
O
Catalysed alternative route
Step 1 4Mn
2+
+ MnO
4
-
+ 8 H
+
5Mn
3
+ + 4 H
2
O
Step 2 2Mn
3+
+ C
2
O
4
2-
2Mn
2+
+ 2 CO
2
The initial uncatalysed reaction is slow because the reaction is a
collision between two negative ions which repel each other
leading to a high activation energy.
The Mn
2+
ions produced act as an autocatalyst and therefore the
reaction starts to speed up because they bring about the
alternative reaction route with lower activation energy.
The reaction eventually slows as the MnO
4
-
concentration drops.
This is an example of autocatalysis where one of
the products of the reaction can catalyse the
reaction.
Mixture with only
reactants showing
effect of autocatalysis
Mixture with
added catalyst
Mn
2+
at start
Alternative method for following the reaction rate
This experiment can be done by removing samples at set times and titrating to work out the concentration of MnO
4
-
.
It could also be done by use of a spectrometer measuring the intensity of the purple colour. This method has the
advantage that it does not disrupt the reaction mixture, using up the reactants and it leads to a much quicker
determination of concentration.
Autocatalytic Reaction between Ethanedioate and Manganate ions
Explanation of results
